
BMS-986115
CAS 1584647-27-7
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide
MW: 574.4945, C26-H25-F7-N4-O3, UNII: LSK1L593UU
10-Nitrooleate, CTK3B7458, CTK3C3167, 9-Octadecenoic acid, 10-nitro-, 875685-46-4, AG-L-63109, 9-Octadecenoic acid, 10-nitro-, (9E)-, 88127-53-1
FOR advanced solid tumors
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- Mechanism of Action Amyloid precursor protein secretase inhibitors; Notch signalling pathway inhibitors
- Phase I Solid tumours
Most Recent Events
- 30 Aug 2016Bristol-Myers Squibb terminates a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA, Australia and Canada (NCT01986218)
- 25 Jan 2016Bristol-Myers Squibb completes enrolment in its phase I trial for Solid tumours in USA, Australia and Canada (NCT01986218)
- 31 Dec 2013Phase-I clinical trials in Solid tumours (late-stage disease) in Canada & Australia (Oral)
DETAILS WILL BE UPDATED SOON………….
BMS-986115 is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, GS/pan-Notch inhibitor BMS 986115 binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival
| Ashvinikumar V. Gavai, George V. Delucca,Daniel O’MALLEY, Patrice Gill, Claude A. Quesnelle, Brian E. Fink, Yufen Zhao,Francis Y. Lee, | |
| Applicant | Bristol-Myers Squibb Company |

Ashvinikumar Gavai

Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb

RICHARD LEE


Patrice Gill
Research scientist at BMS

Dan O’Malley (Rice University)
Currently: Bristol-Myers Squibb
PICTURES WILL BE UPDATED………….
useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()

PATENTS
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en


Scheme 3


XII XI
Scheme 4

Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate

[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate

[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
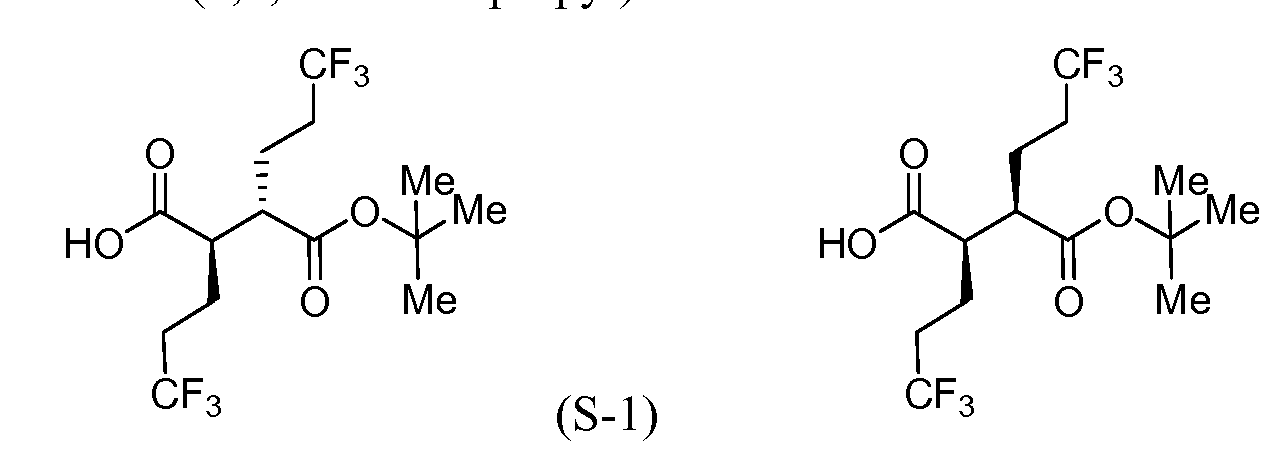
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate

[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one

[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate

[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid

Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate

[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid

[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide

[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one

Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate

(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide

Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat

[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors

Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
PATENT RELATED






PATENTS RELATED
Clip RELATED
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.

Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.

(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html

PAPER RELATED

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, UNII: LSK1L593UU
Cc1cccc2c1NC(=O)[C@H](N=C2c3cccc(c3)F)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N
Filed under: Uncategorized Tagged: BMS-986115







