
ACT-334441
Cenerimod
UNII-Y333RS1786; Y333RS1786
S1P receptor 1 agonist
CAS 1262414-04-9
Chemical Formula: C25H31N3O5
Exact Mass: 453.22637
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
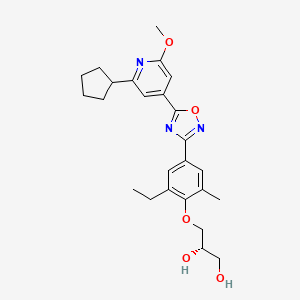
(S)-3-(4-(5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl)-2-ethyl-6-methylphenoxy)propane-1,2-diol
(2S)-3-[4-[5-(2-cyclopentyl-6-methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl]-2-ethyl-6-methylphenoxy]propane-1,2-diol
(S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
| Mechanism Of Action | Sphingosine 1 phosphate receptor modulator |
|---|---|
| Who Atc Codes | L03A-X (Other immunostimulants) |
| Ephmra Codes | L3A (Immunostimulating Agents Excluding Interferons) |
| Indication | Systemic Lupus Erythematosus |
Cenerimod is a potent and orally active immunomodulator, exhibited EC50 value of 2.7 nM. Cenerimod is an agonist for the G protein-coupled receptor S1 P1/EDG1 and has a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. Cenerimod may be useful for prevention or treatment of diseases associated with an activated immune system

CENERIMOD
ACT-334441; lysosphingolipid receptor agonist – Actelion; S1P1 receptor modulator – Actelion; Second selective S1P1 receptor agonist – Actelion; Sphingosine 1 phosphate receptor modulators – Actelion; Sphingosine 1-phosphate receptor 1 agonists – Actelion
- Mechanism of Action Lysosphingolipid receptor agonists
- Highest Development Phases
- Phase I/II Systemic lupus erythematosus
Most Recent Events
- 09 Jun 2016 Actelion terminates a phase I drug interaction trial for Systemic lupus erythematosus (In volunteers) in France (NCT02479204)
- 22 Dec 2015 Phase-I/II clinical trials in Systemic lupus erythematosus in Ukraine, Belarus (PO) (NCT02472795)
- 24 Sep 2015 Phase-I/II clinical trials in Systemic lupus erythematosus in USA (PO) (NCT02472795)
| # | Nct Number | Title | Recruitment | Conditions | Interventions | Phase | |
|---|---|---|---|---|---|---|---|
| 1 | NCT02472795 | Clinical Study to Investigate the Biological Activity, Safety, Tolerability, and Pharmacokinetics of ACT-334441 in Subjects With Systemic Lupus Erythematosus | Recruiting | Systemic Lupus Erythematosus | Drug: ACT-334441|Drug: Placebo | Phase 2 Actelion | |
| 2 | NCT02479204 | Drug Interaction Study of ACT-334441 With Cardiovascular Medications in Healthy Subjects | Suspended | Healthy Subjects | Drug: ACT-334441 2 mg|Drug: ACT-334441 4 mg|Drug: placebo|Drug: atenolol|Drug: diltiazem ER | Phase 1 Actelion |



The human immune system is designed to defend the body against foreign micro-organisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
SYNTHESIS

PATENT
https://www.google.com/patents/WO2011007324A1?cl=zh
The human immune system is designed to defend the body against foreign microorganisms and substances that cause infection or disease. Complex regulatory mechanisms ensure that the immune response is targeted against the intruding substance or organism and not against the host. In some cases, these control mechanisms are unregulated and autoimmune responses can develop. A consequence of the uncontrolled inflammatory response is severe organ, cell, tissue or joint damage. With current treatment, the whole immune system is usually suppressed and the body’s ability to react to infections is also severely compromised. Typical drugs in this class include azathioprine, chlorambucil, cyclophosphamide, cyclosporin, or methotrexate. Corticosteroids which reduce inflammation and suppress the immune response, may cause side effects when used in long term treatment. Nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce pain and inflammation, however, they exhibit considerable side effects. Alternative treatments include agents that activate or block cytokine signaling.
Orally active compounds with immunomodulating properties, without compromising immune responses and with reduced side effects would significantly improve current treatments of uncontrolled inflammatory diseases.
In the field of organ transplantation the host immune response must be suppressed to prevent organ rejection. Organ transplant recipients can experience some rejection even when they are taking immunosuppressive drugs. Rejection occurs most frequently in the first few weeks after transplantation, but rejection episodes can also happen months or even years after transplantation. Combinations of up to three or four medications are commonly used to give maximum protection against rejection while minimizing side effects. Current standard drugs used to treat the rejection of transplanted organs interfere with discrete intracellular pathways in the activation of T-type or B-type white blood cells. Examples of such drugs are cyclosporin, daclizumab, basiliximab, everolimus, or FK506, which interfere with cytokine release or signaling; azathioprine or leflunomide, which inhibit nucleotide synthesis; or 15-deoxyspergualin, an inhibitor of leukocyte differentiation.
The beneficial effects of broad immunosuppressive therapies relate to their effects; however, the generalized immunosuppression which these drugs produce diminishes the immune system’s defense against infection and malignancies. Furthermore, standard immunosuppressive drugs are often used at high dosages and can cause or accelerate organ damage.
Description of the invention
The present invention provides novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1 P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T- / B-lymphocytes as a result of S1 P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1 P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1 P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371 , which are considered closest prior art analogues are shown in Figure 1.

Figure 1 : Structure of Examples of prior art document WO 2008/029371 , which are considered closest analogues to the compounds of the present invention.
The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371 , respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371 , respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents an isobutyl and R2 represents methoxy or wherein R1represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1 is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371 , respectively. This proves that compounds wherein R1represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371 , i.e. the compounds wherein R1 represents methyl and R2 represents cyclopentyl.
Also, the compound of Example 3 of the present invention exhibits the same low potential to constrict rat trachea rings as its S-enantiomer, i.e. the compound of Example 2 of the present invention, indicating that the configuration at this position has no significant effect on trachea constriction. Furthermore, also Example 21 of the present invention exhibits the same low potential to constrict rat trachea rings as present Example 2, which differs from Example 21 only by the linker A (forming a 5-pyridin-4-yl-[1 ,2,4]oxadiazole instead of a 3- pyridin-4-yl-[1 ,2,4]oxadiazole). This indicates that also the nature of the oxadiazole is not critical regarding trachea constriction.
Table 1 : Rat trachea constriction in % of the constriction induced by 50 mM KCI. n.d. = not determined. For experimental details and further data see Example 33.


result obtained at a compound concentration of 300 nM.
The compounds of the present invention can be utilized alone or in combination with standard drugs inhibiting T-cell activation, to provide a new immunomodulating therapy with a reduced propensity for infections when compared to standard immunosuppressive therapy. Furthermore, the compounds of the present invention can be used in combination with reduced dosages of traditional immunosuppressant therapies, to provide on the one hand effective immunomodulating activity, while on the other hand reducing end organ damage associated with higher doses of standard immunosuppressive drugs. The observation of improved endothelial cell layer function associated with S1 P1/EDG1 activation provides additional benefits of compounds to improve vascular function.
The nucleotide sequence and the amino acid sequence for the human S1 P1/EDG1 receptor are known in the art and are published in e.g.: HIa, T., and Maciag, T., J. Biol
Chem. 265 (1990), 9308-9313; WO 91/15583 published 17 October 1991 ; WO 99/46277 published 16 September 1999. The potency and efficacy of the compounds of Formula (I) are assessed using a GTPγS assay to determine EC5O values and by measuring the circulating lymphocytes in the rat after oral administration, respectively (see in experimental part). i) In a first embodiment, the invention relates to pyridine compounds of the Formula (I),

Formula (I)
PATENT
WO 2013175397
https://www.google.com/patents/WO2013175397A1?cl=en
Pyridine-4-yl derivatives of formula (PD),

Formula (PD) A represents

(the asterisks indicate the bond that is linked to the pyridine group of Formula (PD));
Ra represents 3-pentyl, 3-methyl-but-1-yl, cyclopentyl, or cyclohexyl;
Rb represents methoxy;
Rc represents 2,3-dihydroxypropoxy, -OCH2-CH(OH)-CH2-NHCO-CH2OH,
-OCH2-CH(OH)-CH2N(CH3)-CO-CH2OH, -NHS02CH3, or -NHS02CH2CH3; and
Rd represents ethyl or chloro.)
disclosed in WO201 1007324, have immunomodulating activity through their S1 P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and / or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO201 1007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
In the process described in WO201 1007324, 2-cyclopentyl-6-methoxy-isonicotinic acid was prepared according to the following reaction scheme 1 :

Compound D Compound E
Rieke Zinc: cyclopentylzinc bromide;
PdCI2(dppf)dcm: 1 ,1 ‘-Bis(diphenylphosphino)ferrocene-palladium(ll)dichloride
dichloromethane complex
However, the abovementioned process has drawbacks for larger scale, i.e. industrial scale synthesis of 2-cyclopentyl-6-methoxy-isonicotinic acid, for the following reasons:
a) The commercially available starting material, 2,6-dichloro-isonicotinic acid (Compound A) is expensive.
b) The conversion of Compound C to Compound D is cost-intensive. The reaction has to be performed under protective atmosphere with expensive palladium catalysts and highly reactive and expensive Rieke zinc complex. Such synthesis steps are expensive to scale up and it was therefore highly desired to find alternative synthesis methods.
Even though Goldsworthy, J. Chem. Soc. 1934, 377-378 discloses the preparation of 1 -cyclopentylethanone, which is a key building block in the new process of the present invention, by using ethyl 1 -acetoacetate as a starting material, this synthesis was far from being suitable in an industrial process. The reported yield was low (see also under “Referential Examples” below). Scheme 2

ethyl 1 -acetylcyclo- 1-cyclopentyl- pentanecarboxylate ethanone
Besides the early work by Goldsworthy there are several recent examples for the preparation of 1 -cyclopentylethanone described in the literature. Such examples include:
1 ) Addition of methyl lithium to a N-cyclopentanecarbonyl-N,0-dimethylhydroxylamine at -78°C in a yield of 77%. US2006/199853 A1 , 2006 and US2006/223884 A1 , 2006.
2) Addition of methyl lithium to a cyclopentyl carboxylic acid in diethylether at -78°C in a yield of 81 %. J. Am. Chem. Soc, 1983, 105, 4008-4017.
3) Addition of methylmagnesiumbromide to cyclopentanecarbonitrile.
Bull. Soc. Chim. Fr., 1967, 3722-3729.
4) Oxidation of 1 -cyclopentylethanol with chromtrioxide. US5001 140 A1 , 1991.
WO2009/71707 A1 , 2009.
5) Addition of cyclopentylmagnesium bromide to acetic anhydride at -78 °C with a yield of 54%. WO2004/74270 A2, 2004.
6) Synthesis of 1-cyclopentylethanone in 5 steps from cyclopentanone. Zhang, Pang; Li, Lian-chu, Synth. Commun., 1986, 16, 957-966.
However, the processes described in the above-listed publications are not efficient for scale-up since they require cryogenic temperatures, expensive starting materials, toxic reagents or many steps. The lack of an efficient process to manufacture 1 -cyclopentylethanone is further also mirrored by the difficulty in sourcing this compound on kilogram scale for a reasonable price and delivery time. Therefore, the purpose of the present invention is to provide a new, efficient and cost effective process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is suitable for industrial scale synthesis.
Patent
Disclosed in WO2011007324, have immunomodulating activity through their S1P1/EDG1 receptor agonistic activity. Therefore, those pyridine-4-yl derivatives are useful for prevention and/or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto’s thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis. 2-Cyclopentyl-6-methoxy-isonicotinic acid, which is also disclosed in WO2011007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein Ra is a cyclopentyl group.
 Rieke Zinc: cyclopentylzinc bromide;
Rieke Zinc: cyclopentylzinc bromide;
PdCl2(dppf)dcm: 1,1′-Bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex


EXAMPLES
Example 1a
1-Cyclopentylethanone

Example 1 b
1-Cyclopentylethanone
Example 1c
1-Cyclopentylethanone
Example 1d
1-Cyclopentylethanone
Example 1e
1-Cyclopentylethanone
Example 1f
Tert-butyl 1-acetylcyclopentanecarboxylate

Example 1 g
Tert-butyl 1-acetylcyclopentanecarboxylate
Example 2
2-Cyclopentyl-6-hydroxyisonicotinic acid

Example 3
Methyl 2-cyclopentyl-6-hydroxyisonicotinate

Example 4a
Methyl 2-chloro-6-cyclopentylisonicotinate

Example 4b
Methyl 2-chloro-6-cyclopentylisonicotinate
Example 5
2-Cyclopentyl-6-methoxyisonicotinic acid

Example 6
Ethyl 4-cyclopentyl-2,4-dioxobutanoate

Example 7
Ethyl 3-cyano-6-cyclopentyl-2-hydroxyisonicotinate

REFERENTIAL EXAMPLES

Referential Example 1
Referential Example 2
PATENT
https://www.google.com/patents/US8658675
Martin Bolli, Cyrille Lescop, Boris Mathys,Keith Morrison, Claus Mueller, Oliver Nayler,Beat Steiner,
novel compounds of Formula (I) that are agonists for the G protein-coupled receptor S1P1/EDG1 and have a powerful and long-lasting immunomodulating effect which is achieved by reducing the number of circulating and infiltrating T- and B-lymphocytes, without affecting their maturation, memory, or expansion. The reduction of circulating T-/B-lymphocytes as a result of S1P1/EDG1 agonism, possibly in combination with the observed improvement of endothelial cell layer function associated with S1P1/EDG1 activation, makes such compounds useful to treat uncontrolled inflammatory diseases and to improve vascular functionality. Prior art document WO 2008/029371 discloses compounds that act as S1P1/EDG1 receptor agonists and show an immunomodulating effect as described above. Unexpectedly, it has been found that the compounds of the present invention have a reduced potential to constrict airway tissue/vessels when compared to compounds of the prior art document WO 2008/029371. The compounds of the present invention therefore demonstrate superiority with respect to their safety profile, e.g. a lower risk of bronchoconstriction.
Examples of WO 2008/029371, which are considered closest prior art analogues are shown in FIG. 1.


The data on the constriction of rat trachea rings compiled in Table 1 illustrate the superiority of the compounds of the present invention as compared to compounds of prior art document WO 2008/029371.
For instance, the compounds of Example 1 and 6 of the present invention show a significantly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 222 and 226 of WO 2008/029371, respectively. Furthermore, the compounds of Example 1 and 6 of the present invention also show a reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 196 and 204 of WO 2008/029371, respectively. These data demonstrate that compounds wherein R1 represents 3-pentyl and R2represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1 represents an isobutyl and R2represents methoxy or wherein R1 represents methyl and R2 represents 3-pentyl. Moreover, also the compound of Example 16 of the present invention, wherein R1is 3-methyl-but-1-yl and R2 is methoxy, exhibits a markedly reduced potential to constrict rat trachea rings when compared to its closest analogue prior art Example 226 of WO 2008/029371 wherein R1 is isobutyl and R2 is methoxy.
The unexpected superiority of the compounds of the present invention is also evident from the observation that the compounds of Example 2 and 7 of the present invention show a markedly reduced potential to constrict rat trachea rings when compared to the compounds of prior art Examples 229 and 233 of WO 2008/029371, respectively. This proves that compounds wherein R1 represents cyclopentyl and R2 represents methoxy are superior compared to the closest prior art compounds of WO 2008/029371, i.e. the compounds wherein R1represents methyl and R2 represents cyclopentyl.
Preparation of Intermediates2-Chloro-6-methyl-isonicotinic acid
The title compound and its ethyl ester are commercially available.
2-(1-Ethyl-propyl)-6-methoxy-isonicotinic acid
a) To a solution of 2,6-dichloroisonicotinic acid (200 g, 1.04 mol) in methanol (3 L), 32% aq. NaOH (770 mL) is added. The stirred mixture becomes warm (34° C.) and is then heated to 70° C. for 4 h before it is cooled to rt. The mixture is neutralised by adding 32% aq. HCl (100 mL) and 25% aq. HCl (700 mL). The mixture is stirred at rt overnight. The white precipitate that forms is collected, washed with methanol and dried. The filtrate is evaporated and the residue is suspended in water (200 mL). The resulting mixture is heated to 60° C. Solid material is collected, washed with water and dried. The combined crops give 2-chloro-6-methoxy-isonicotinic acid (183 g) as a white solid; LC-MS: tR=0.80 min, [M+1]+=187.93.
b) To a suspension of 2-chloro-6-methoxy-isonicotinic acid (244 g, 1.30 mol) in methanol (2.5 L), H2SO4 (20 mL) is added. The mixture is stirred at reflux for 24 h before it is cooled to 0° C. The solid material is collected, washed with methanol (200 mL) and water (500 mL) and dried under HV to give 2-chloro-6-methoxy-isonicotinic acid methyl ester (165 g) as a white solid; LC-MS: tR=0.94 min, [M+1]+=201.89.
c) Under argon, Pd(dppf) (3.04 g, 4 mmol) is added to a solution of 2-chloro-6-methoxy-isonicotinic acid methyl ester (50 g, 0.248 mol) in THF (100 mL). A 0.5 M solution of 3-pentylzincbromide in THF (550 mL) is added via dropping funnel. Upon complete addition, the mixture is heated to 85° C. for 18 h before it is cooled to rt. Water (5 mL) is added and the mixture is concentrated. The crude product is purified by filtration over silica gel (350 g) using heptane:EA 7:3 to give 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (53 g) as a pale yellow oil; 1H NMR (CDCl3): δ0.79 (t, J=7.5 Hz, 6H), 1.63-1.81 (m, 4H), 2.47-2.56 (m, 1H), 3.94 (s, 3H), 3.96 (s, 3H), 7.12 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
d) A solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid methyl ester (50 g, 0.211 mol) in ethanol (250 mL), water (50 mL) and 32% aq. NaOH (50 mL) is stirred at 80° C. for 1 h. The mixture is concentrated and the residue is dissolved in water (200 mL) and extracted with TBME. The org. phase is separated and washed once with water (200 mL). The TBME phase is discarded. The combined aq. phases are acidified by adding 25% aq. HCl and then extracted with EA (400+200 mL). The combined org. extracts are concentrated. Water (550 mL) is added to the remaining residue. The mixture is heated to 70° C., cooled to rt and the precipitate that forms is collected and dried to give the title compound (40.2 g) as a white solid; LC-MS: tR=0.95 min, [M+1]+=224.04; 1H NMR (D6-DMSO): δ 0.73 (t, J=7.3 Hz, 6H), 1.59-1.72 (m, 4H), 2.52-2.58 (m, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H).
2-Methoxy-6-(3-methyl-butyl)-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.94 min, [M+1]+=224.05; 1H NMR (D6-DMSO): δ 0.92 (d, J=5.8 Hz, 6H), 1.54-1.62 (m, 3H), 2.70-2.76 (m, 2H), 3.88 (s, 3H), 6.99 (s, 1H), 7.25 (s, 1H), 13.52 (s).
2-Cyclopentyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.93 min, [M+1]+=222.02; 1H NMR (CDCl3): δ 1.68-1.77 (m, 2H), 1.81-1.90 (m, 4H), 2.03-2.12 (m, 2H), 3.15-3.25 (m, 1H), 3.99 (s, 3H), 7.18 (d, J=1.0 Hz, 1H), 7.35 (d, J=0.8 Hz, 1H).
2-Cyclohexyl-6-methoxy-isonicotinic acid
The title compound is prepared in analogy to 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid; LC-MS: tR=0.98 min, [M+1]+=236.01; 1H NMR (D6-DMSO): δ 1.17-1.29 (m, 1H), 1.31-1.43 (m, 2H), 1.44-1.55 (m, 2H), 1.67-1.73 (m, 1H), 1.76-1.83 (m, 2H), 1.84-1.92 (m, 2H), 2.66 (tt, J=11.3, 3.3 Hz, 1H), 3.88 (s, 3H), 7.00 (d, J=1.0 Hz, 1H), 7.23 (d, J=1.0 Hz, 1H).
2-Cyclopentyl-N-hydroxy-6-methoxy-isonicotinamidine
a) A solution of 2-cyclopentyl-6-methoxy-isonicotinic acid methyl ester (3.19 g, 13.6 mmol) in 7 N NH3 in methanol (50 mL) is stirred at 60° C. for 18 h. The solvent is removed in vacuo and the residue is dried under HV to give crude 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g) as a pale yellow solid; LC-MS**: tR=0.57 min, [M+1]+=221.38.
b) Pyridine (8.86 g, 91.3 mmol) is added to a solution of 2-cyclopentyl-6-methoxy-isonicotinamide (3.35 g, 15.2 mmol) in DCM (100 mL). The mixture is cooled to 0° C. before trifluoroacetic acid anhydride (9.58 g, 45.6 mmol) is added portionwise. The mixture is stirred at 0° C. for 1 h before it is diluted with DCM (100 mL) and washed with sat. aq. NaHCO3 solution (100 mL) and brine (100 mL). The separated org. phase is dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 9:1 to give 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g) as pale yellow oil; LC-MS**: tR=0.80 min, [M+1]+=not detectable; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.16 (quint, J=7.8 Hz, 1H), 3.89 (s, 3H), 7.15 (s, 1H), 7.28 (s, 1H).
c) To a solution of 2-cyclopentyl-6-methoxy-isonicotinonitrile (2.09 g, 10.3 mmol) in methanol (100 mL), hydroxylamine hydrochloride (2.15 g, 31.0 mmol) and NaHCO3 (3.04 g, 36.2 mmol) are added. The mixture is stirred at 60° C. for 18 h before it is filtered and the filtrate is concentrated. The residue is dissolved in EA (300 mL) and washed with water (30 mL). The washings are extracted back with EA (4×100 mL) and DCM (4×100 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried under HV to give the title compound (2.74 g) as a white solid; LC-MS**: tR=0.47 min, [M+1]+=236.24; 1H NMR (D6-DMSO): δ 1.61-1.82 (m, 6H), 1.92-2.01 (m, 2H), 3.04-3.13 (m, 1H), 3.84 (s, 3H), 5.90 (s, 2H), 6.86 (s, 1H), 7.13 (s, 1H), 9.91 (s, 1H).
2-Cyclopentyl-6-methoxy-isonicotinic acid hydrazide
a) To a solution of 2-cyclopentyl-6-methoxy-isonicotinic acid (2.00 g, 9.04 mmol), hydrazinecarboxylic acid benzyl ester (1.50 g, 9.04 mmol) and DIPEA (2.34 g, 18.1 mmol) in DCM (40 mL), TBTU (3.19 g, 9.94 mmol) is added. The mixture is stirred at rt for 2 h before it is diluted with EA (250 mL), washed twice with sat. aq. NaHCO3 solution (150 mL) followed by brine (100 mL), dried over MgSO4, filtered and concentrated. The crude product is purified by CC on silica gel eluting with heptane:EA 4:1 to give N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g) as pale yellow oil; LC-MS**: tR=0.74 min, [M+1]+=369.69; 1H NMR (D6-DMSO): δ 1.62-1.83 (m, 6H), 1.95-2.05 (m, 2H), 3.10-3.21 (m, 1H), 3.88 (s, 3H), 5.13 (s, 2H), 6.97 (s, 1H), 7.23 (s, 1H), 7.28-7.40 (m, 5H), 9.45 (s, 1H), 10.52 (s, 1H).
b) Pd/C (500 mg, 10% Pd) is added to a solution of N′-(2-cyclopentyl-6-methoxy-pyridine-4-carbonyl)-hydrazinecarboxylic acid benzyl ester (2.74 g, 7.42 mmol) in THF (50 mL) and methanol (50 mL). The mixture is stirred at rt under 1 bar of H2 for 25 h. The catalyst is removed by filtration and the filtrate is concentrated and dried under HV to give the title compound (1.58 g) as an off-white solid; LC-MS**: tR=0.51 min, [M+1]+=236.20; 1H NMR (D6-DMSO): δ 1.60-1.82 (m, 6H), 1.94-2.03 (m, 2H), 3.08-3.19 (m, 1H), 3.86 (s, 3H), 4.56 (s br, 2H), 6.93 (d, J=1.0 Hz, 1H), 7.20 (d, J=1.0 Hz, 1H), 9.94 (s, 1H).
3-Ethyl-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzaldehyde following literature procedures (A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268); LC-MS: tR=0.90 min; 1H NMR (CDCl3): δ1.24 (t, J=7.6 Hz, 3H), 2.26 (s, 3H), 2.63 (q, J=7.6 Hz, 2H), 5.19 (s, 1H), 7.30 (s, 2H).
3-Chloro-4-hydroxy-5-methyl-benzonitrile
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (see 3-ethyl-4-hydroxy-5-methyl-benzonitrile); LC-MS: tR=0.85 min. 1H NMR (CDCl3): δ2.33 (s, 3H), 6.10 (s, 1H), 7.38 (s, 1H), 7.53 (d, J=1.8 Hz, 1H).
3-Ethyl-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from 3-ethyl-4-hydroxy-5-methyl-benzonitrile or from commercially available 2-ethyl-6-methyl-phenol following literature procedures (G. Trapani, A. Latrofa, M. Franco, C. Altomare, E. Sanna, M. Usala, G. Biggio, G. Liso, J. Med. Chem. 41 (1998) 1846-1854; A. K. Chakraborti, G. Kaur, Tetrahedron 55 (1999) 13265-13268; E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.55 min; 1H NMR (D6-DMSO): δ 9.25 (s br, 1H), 7.21 (s, 2H), 5.56 (s, 2H), 2.55 (q, J=7.6 Hz, 2H), 2.15 (s, 3H), 1.10 (t, J=7.6 Hz, 3H).
3-Chloro-4,N-dihydroxy-5-methyl-benzamidine
The title compound is prepared from commercially available 2-chloro-6-methyl-phenol in analogy to literature procedures (e.g. B. Roth et al. J. Med. Chem. 31 (1988) 122-129; and literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine); 3-chloro-4-hydroxy-5-methyl-benzaldehyde: LC-MS: tR=0.49 min, [M+1]+=201.00; 1H NMR 82.24 (s, 2H), 2.35 (s, 4H), 5.98 (s br, 1H), 7.59 (d, J=1.8 Hz, 1H), 7.73 (d, J=1.8 Hz, 1H), 9.80 (s, 1H); 3-chloro-4,N-dihydroxy-5-methyl-benzamidine: 1H NMR (D6-DMSO): δ 2.21 (s, 3H), 5.72 (s br, 2H), 7.40 (s, 1H), 7.48 (s, 1H), 9.29 (s br, 1H), 9.48 (s br, 1H).
(R)-4-(2,2-Dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (2.89 g, 17.9 mmol) in THF (80 mL), (R)-(2,2-dimethyl-[1,3]dioxolan-4-yl)methanol (2.84 g, 21.5 mmol) followed by triphenylphosphine (5.81 g, 21.5 mmol) is added. The mixture is cooled with an ice-bath before DEAD (9.36 g, 21.5 mmol) is added dropwise. The mixture is stirred at rt for 1 h, the solvent is removed in vacuo and the residue is purified by CC on silica gel eluting with heptane:EA 85:15 to give (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g) as a pale yellow oil; LC-MS**: tR=0.75 min, [M+1]+=not detected; 1H NMR (CDCl3): δ1.25 (t, J=7.5 Hz, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 2.34 (s, 3H), 2.65-2.77 (m, 2H), 3.80-3.90 (m, 2H), 3.94-4.00 (m, 1H), 4.21 (t, J=7.3 Hz, 1H), 4.52 (quint, J=5.8 Hz, 1H), 7.35 (s, 1H), 7.38 (s, 1H).
b) To a mixture of (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-benzonitrile (4.45 g, 16.2 mmol) and NaHCO3 (4.75 g, 56.6 mmol) in methanol (30 mL), hydroxylamine hydrochloride (3.37 g, 48.5 mmol) is added. The mixture is stirred at 60° C. for 18 h before it is filtered and the solvent of the filtrate is removed in vacuo. The residue is dissolved in EA and washed with a small amount of water and brine. The org. phase is separated, dried over MgSO4, filtered, concentrated and dried to give the title compound (5.38 g) as a white solid; LC-MS**: tR=0.46 min, [M+1]+=309.23; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 1.33 (s, 3H), 1.38 (s, 3H), 2.25 (s, 3H), 2.57-2.69 (m, 2H), 3.73-3.84 (m, 3H), 4.12 (t, J=7.0 Hz, 1H), 4.39-4.45 (m, 1H), 5.76 (s br, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 9.47 (s, 1H).
(R)-3-Chloro-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-N-hydroxy-5-methyl-benzamidine
The title compound is obtained as a colorless oil (1.39 g) in analogy to (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile and L-α,β-isopropyliden glycerol; LC-MS: tR=0.66 min, [M+H]+=314.96.
(S)-4-(3-Amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzonitrile (5.06 g, 31.4 mmol) in THF (80 mL), PPh3 (9.06 g, 34.5 mmol) and (R)-glycidol (2.29 mL, 34.5 mmol) are added. The mixture is cooled to 0° C. before DEAD in toluene (15.8 mL, 34.5 mmol) is added. The mixture is stirred for 18 h while warming up to rt. The solvent is evaporated and the crude product is purified by CC on silica gel eluting with heptane:EA 7:3 to give 3-ethyl-5-methyl-4-oxiranylmethoxy-benzonitrile (5.85 g) as a yellow oil; LC-MS: tR=0.96 min; [M+42]+=259.08.
b) The above epoxide is dissolved in 7 N NH3 in methanol (250 mL) and the solution is stirred at 65° C. for 18 h. The solvent is evaporated to give crude (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g) as a yellow oil; LC-MS: tR=0.66 min; [M+1]+=235.11.
N—((S)-3-[2-Ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
a) To a solution of (S)-4-(3-amino-2-hydroxypropoxy)-3-ethyl-5-methylbenzonitrile (6.23 g, 26.59 mmol) in THF (150 mL), glycolic acid (2.43 g, 31.9 mmol), HOBt (4.31 g, 31.9 mmol), and EDC hydrochloride (6.12 g, 31.9 mmol) are added. The mixture is stirred at rt for 18 h before it is diluted with sat. aq. NaHCO3 and extracted twice with EA. The combined org. extracts are dried over MgSO4, filtered and concentrated. The crude product is purified by CC with DCM containing 8% of methanol to give (S)—N-[3-(4-cyano-2-ethyl-6-methyl-phenoxy)-2-hydroxy-propyl]-2-hydroxy-acetamide (7.03 g) as a yellow oil; LC-MS: tR=0.74 min, [M+1]+=293.10; 1H NMR (CDCl3): δ 1.25 (t, J=7.5 Hz, 3H), 2.32 (s, 3H), 2.69 (q, J=7.5 Hz, 2H), 3.48-3.56 (m, 3H), 3.70-3.90 (m, 3H), 4.19 (s, br, 3H), 7.06 (m, 1H), 7.36 (s, 1H), 7.38 (s, 1H).
b) The above nitrile is converted to the N-hydroxy-benzamidine according to literature procedures (e.g. E. Meyer, A. C. Joussef, H. Gallardo, Synthesis 2003, 899-905); LC-MS: tR=0.51 min, [M+1]+=326.13; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.4 Hz, 3H), 2.24 (s, 3H), 2.62 (q, J=7.4 Hz, 2H), 3.23 (m, 1H), 3.43 (m, 1H), 3.67 (m, 2H), 3.83 (s, 2H), 3.93 (m, 1H), 5.27 (s br, 1H), 5.58 (s br, 1H), 5.70 (s, 2H), 7.34 (s, 1H), 7.36 (s, 1H), 7.67 (m, 1H), 9.46 (s br, 1H).
(S)—N-(3-[2-Chloro-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide
The title compound is obtained as a beige wax (1.1 g) in analogy to N—((S)-3-[2-ethyl-4-(N-hydroxycarbamimidoyl)-6-methyl-phenoxy]-2-hydroxy-propyl)-2-hydroxy-acetamide starting from 3-chloro-4-hydroxy-5-methyl-benzonitrile; LC-MS: tR=0.48 min, [M+H]+=331.94.
3-Chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) A mixture of 4-amino-3-chloro-5-methylbenzonitrile (155 mg, 930 μmol) and methanesulfonylchloride (2.13 g, 18.6 mmol, 1.44 mL) is heated under microwave conditions to 150° C. for 7 h. The mixture is cooled to rt, diluted with water and extracted with EA. The org. extract is dried over MgSO4, filtered and concentrated. The crude product is purified on prep. TLC using heptane:EA 1:1 to give N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg) as an orange solid; LC-MS**: tR=0.48 min; 1H NMR (CDCl3): δ2.59 (s, 3H), 3.18 (s, 3H), 6.27 (s, 1H), 7.55 (d, J=1.3 Hz, 1H), 7.65 (d, J=1.5 Hz, 1H).
b) Hydroxylamine hydrochloride (60 mg, 858 μmol) and NaHCO3 (72 mg, 858 μmol) is added to a solution of N-(2-chloro-4-cyano-6-methyl-phenyl)-methanesulfonamide (105 mg, 429 μmol) in methanol (10 mL). The mixture is stirred at 65° C. for 18 h. The solvent is removed in vacuo and the residue is dissolved in a small volume of water (2 mL) and extracted three times with EA (15 mL). The combined org. extracts are dried over MgSO4, filtered, concentrated and dried to give the title compound (118 mg) as a white solid; LC-MS**: tR=0.19 min, [M+1]+=277.94; 1H NMR (CDCl3): δ2.57 (s, 3H), 3.13 (s, 3H), 6.21 (s, 1H), 7.49 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz).
3-Ethyl-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine
a) In a 2.5 L three-necked round-bottom flask 2-ethyl-6-methyl aniline (250 g, 1.85 mol) is dissolved in DCM (900 mL) and cooled to 5-10° C. Bromine (310.3 g, 1.94 mol) is added over a period of 105 min such as to keep the temperature at 5-15° C. An aq. 32% NaOH solution (275 mL) is added over a period of 10 min to the greenish-grey suspension while keeping the temperature of the reaction mixture below 25° C. DCM (70 mL) and water (100 mL) are added and the phases are separated. The aq. phase is extracted with DCM (250 mL). The combined org. phases are washed with water (300 mL) and concentrated at 50° C. to afford the 4-bromo-2-ethyl-6-methyl-aniline (389 g) as a brown oil; 1H NMR (CDCl3): δ 1.27 (t, J=7.3 Hz, 3H), 2.18 (s, 3H), 2.51 (q, J=7.3 Hz, 2H), 3.61 (s br, 1H), 7.09 (s, 2H).
b) A double-jacketed 4 L-flask is charged with 4-bromo-2-ethyl-6-methyl-aniline (324 g, 1.51 mol), sodium cyanide (100.3 g, 1.97 mol), potassium iodide (50.2 g, 0.302 mol) and copper(I)iodide (28.7 g, 0.151 mol). The flask is evacuated three times and refilled with nitrogen. A solution of N,N′-dimethylethylenediamine (191.5 mL, 1.51 mol) in toluene (750 mL) is added. The mixture is heated to 118° C. and stirred at this temperature for 21 h. The mixture is cooled to 93° C. and water (1250 mL) is added to obtain a solution. Ethyl acetate (1250 mL) is added at 22-45° C. and the layers are separated. The org. phase is washed with 10% aq. citric acid (2×500 mL) and water (500 mL). The separated org. phase is evaporated to dryness to afford 4-amino-3-ethyl-5-methyl-benzonitrile (240 g) as a metallic black solid; 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.19 (s, 3H), 2.52 (q, J=7.3 Hz, 2H), 4.10 (s br, 1H), 7.25 (s, 2H).
c) The title compound is then prepared from the above 4-amino-3-ethyl-5-methyl-benzonitrile in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine; LC-MS**: tR=0.26 min, [M+1]+=272.32.
3-Chloro-4-ethanesulfonylamino N-hydroxy-5-methyl-benzamidine
The title compound is prepared in analogy to 3-chloro-N-hydroxy-4-methanesulfonylamino-5-methyl-benzamidine using ethanesulfonylchloride; LC-MS**: tR=0.27 min, [M+1]+=292.13; 1H NMR (D6-DMSO): δ 1.36 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 3.22 (q, J=7.5 Hz), 5.88 (s, 2H), 7.57 (d, J=1.5 Hz, 1H), 7.63 (d, J=1.5 Hz, 1H), 9.18 (s, 1H), 9.78 (s, 1H).
4-Benzyloxy-3-ethyl-5-methyl-benzoic acid
a) To a solution of 3-ethyl-4-hydroxy-5-methyl-benzaldehyde (34.9 g, 0.213 mol, prepared from 2-ethyl-6-methyl-phenol according to the literature cited for 3-ethyl-4,N-dihydroxy-5-methyl-benzamidine) in MeCN (350 mL), K2CO3 (58.7 g, 0.425 mol) and benzylbromide (36.4 g, 0.213 mol) are added. The mixture is stirred at 60° C. for 2 h before it is cooled to rt, diluted with water and extracted twice with EA. The org. extracts are washed with water and concentrated to give crude 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (45 g) as an orange oil. 1H NMR (CDCl3): δ1.29 (t, J=7.5 Hz, 3H), 2.40 (s, 3H), 2.77 (q, J=7.8 Hz, 2H), 4.90 (s, 2H), 7.31-7.52 (m, 5H), 7.62 (d, J=1.5 Hz, 1H), 7.66 (d, J=1.8 Hz, 1H), 9.94 (s, 1H).
b) To a mixture of 4-benzyloxy-3-ethyl-5-methyl-benzaldehyde (132 g, 0.519 mol) and 2-methyl-2-butene (364 g, 5.19 mol) in tert.-butanol (1500 mL), a solution of NaH2PO4 dihydrate (249 g, 2.08 mol) in water (1500 mL) is added. To this mixture, NaClO2 (187.8 g, 2.08 mol) is added in portions. The temperature of the reaction mixture is kept below 30° C., and evolution of gas is observed. Upon completion of the addition, the orange bi-phasic mixture is stirred well for 3 h before it is diluted with TBME (1500 mL). The org. layer is separated and washed with 20% aq. NaHS solution (1500 mL) and water (500 mL). The org. phase is then extracted three times with 0.5 N aq. NaOH (1000 mL), the aq. phase is acidified with 25% aq. HCl (500 mL) and extracted twice with TBME (1000 mL). These org. extracts are combined and evaporated to dryness to give the title compound; 1H NMR (D6-DMSO): δ 1.17 (t, J=7.5 Hz, 3H), 2.31 (s, 3H), 2.67 (q, J=7.5 Hz, 2H), 4.86 (s, 2H), 7.34-7.53 (m, 5H), 7.68 (s, 2H), 12.70 (s, 1H).
Example 1 (S)-3-(2-Ethyl-4-{5-[2-(1-ethyl-propyl)-6-methoxy-pyridin-4-yl]-[1,2,4]oxadiazol-3-yl}-6-methyl-phenoxy)-propane-1,2-diol
a) To a solution of 2-(1-ethyl-propyl)-6-methoxy-isonicotinic acid (190 mg, 732 μmol) in THF (10 mL) and DMF (2 mL), DIPEA (190 mg, 1.46 mmol) followed by TBTU (235 mg, 732 μmol) is added. The mixture is stirred at rt for 10 min before (R)-4-(2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-N-hydroxy-5-methyl-benzamidine 226 mg, 732 μmol) is added. The mixture is stirred at rt for 1 h before it is diluted with EA and washed with water. The org. phase is separated and concentrated. The remaining residue is dissolved in dioxane (10 mL) and heated to 105° C. for 18 h. The mixture is cooled to rt, concentrated and the crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (256 mg) as a yellow oil; LC-MS: tR=1.28 min, [M+H]+=496.23.
b) A solution of 4-{3-[4-((R)-2,2-dimethyl-[1,3]dioxolan-4-ylmethoxy)-3-ethyl-5-methyl-phenyl]-[1,2,4]oxadiazol-5-yl}-2-(1-ethyl-propyl)-6-methoxy-pyridine (250 mg, 504 μmol) in 4 M HCl in dioxane (10 mL) is stirred at rt for 90 min before it is concentrated. The crude product is purified on prep. TLC plates using DCM containing 10% of methanol to give the title compound (76 mg) as a pale brownish solid; LC-MS: tR=1.12 min, [M+H]+=456.12; 1H NMR (CDCl3): δ0.85 (t, J=7.0 Hz, 6H), 1.33 (t, J=7.0 Hz, 3H), 1.70-1.89 (m, 4H), 2.42 (s, 3H), 2.61-2.71 (m, 1H), 2.78 (q, J=7.3 Hz, 2H), 3.82-4.00 (m, 4H), 4.04 (s, 3H), 4.14-4.21 (m, 1H), 7.34 (s, 1H), 7.46 (s, 1H), 7.86-7.91 (m, 2H).
Example 2 (S)-3-{4-[5-(2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol
The title compound is prepared in analogy to Example 1 starting from 2-cyclopentyl-6-methoxy-isonicotinic acid; LC-MS: tR=1.14 min, [M+H]+=454.16; 1H NMR (CDCl3): δ1.33 (t, J=7.5 Hz, 3H), 1.72-1.78 (m, 2H), 1.85-1.94 (m, 4H), 2.03-2.15 (m, 2H), 2.41 (s, 3H), 2.72 (d, J=5.3 Hz, 1H), 2.77 (q, J=7.5 Hz, 2H), 3.19-3.28 (m, 1H), 3.81-3.94 (m, 2 H), 3.95-3.98 (m, 2H), 4.02 (s, 3H), 4.14-4.21 (m, 1H), 7.31 (d, J=1.3 Hz, 1H), 7.51 (d, J=1.0 Hz, 1H), 7.88 (d, J=1.8 Hz), 7.89 (d, J=2.0 Hz, 1H).
PAPER

A practical synthesis of S1P receptor 1 agonist ACT-334441 (1) through late-stage convergent coupling of two key intermediates is described. The first intermediate is 2-cyclopentyl-6-methoxyisonicotinic acid whose skeleton was built from 1-cyclopentylethanone, ethyl oxalate, and cyanoacetate in a Guareschi–Thorpe reaction in 42% yield over five steps. The second, chiral intermediate, is a phenol ether derived from enantiomerically pure (R)-isopropylidene glycerol ((R)-solketal) and 3-ethyl-4-hydroxy-5-methylbenzonitrile in 71% yield in a one-pot reaction. The overall sequence entails 18 chemical steps with 10 isolated intermediates. All raw materials are cheap and readily available in bulk quantities, the reaction conditions match with standard pilot plant equipment, and the route reproducibly afforded 3–20 kg of 1 in excellent purity and yield for clinical studies.
Practical Synthesis of a S1P Receptor 1 Agonist via a Guareschi–Thorpe Reaction
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015133669 | 2015-05-14 | NEW PROCESS FOR THE PREPARATION OF 2-CYCLOPENTYL-6-METHOXY-ISONICOTINIC ACID |
| US8658675 | 2014-02-25 | Pyridin-4-yl derivatives |
Filed under: Uncategorized Tagged: ACT-334441, ACT334441, Actelion Pharmaceuticals Ltd., Beat Steiner, Boris Mathys, CENERIMOD, Claus Mueller, Cyrille Lescop, Keith Morrison, Martin Bolli, Oliver Nayler, phase 2, S1P receptor 1 agonist, Systemic lupus erythematosus, UNII-Y333RS1786 Y333RS1786







