![Osanetant.png]()
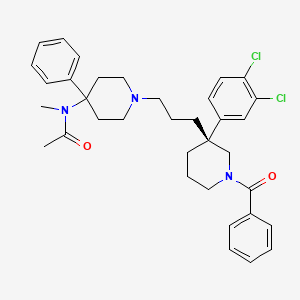
Osanetant (SR-142,801)
160492-56-8 CAS
| : MW 605.257582985 |
| Chemical Formula |
C35H41Cl2N3O2 |
(R)-(+)-N-[[3-[1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide
Osanetant (SR-142,801) was a neurokinin 3 receptor antagonist developed by Sanofi-Synthélabo, which was being researched for the treatment of schizophrenia, but was discontinued.[1][2] It was the first non-peptide NK3 antagonist developed in the mid-1990s,[3][4] Other potential applications for osanetant is in the treatment of drug addiction, as it has been found to block the effects ofcocaine in animal models.[5][6]
Developed by Sanofi-Aventis (formerly Sanofi-Synthelabo), osanetant (SR-142801) is an NK3 receptor antagonist which was under development for the treatment of schizophrenia and other Central Nervous System (CNS) disorders. In a review of its R&D portfolio, the company announced in August 2005 that it would cease any further development ofosanetant. This follows an earlier decision to discontinue development of eplivanserin for schizophrenia
![]()
(R)-(+)-N-[[3-[1-benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide and to a process for their preparation. (R)-(+)-N-[[3-[1-Benzoyl-3-(3,4-dichlorophenyl)piperidin-3-yl]prop-1-yl]-4-phenylpiperidin-4-yl]-N-methylacetamide, hereinafter denoted by its International Non-proprietary Name “osanetant”, is the first antagonist of the NK-3 receptor described in the literature, the preparation of which, in particular in the hydrochloride form, is illustrated in EP-A-673 928.
According to this document, osanetant is prepared by reacting N-methyl-N-(4-phenylpiperidin-4-yl)acetamide with 1-benzoyl-3-(3,4-dichlorophenyl)-3-(methanesulfonyloxyprop-1-yl)piperidine and by converting the osanetant thus obtained to its hydrochloride. It has been found that osanetant hydrochloride is isolated in the form of an amorphous solid which is difficult to purify. This product comprises impurities originating from the preceding synthetic stages.
Preparative chromatography starting from osanetant base can be used to obtain osenetant in the pure form.
Osanetant is a neurokinin (NK3) receptor antagonist under development by Sanofi-Synthélabo (formerly Sanofi) as a potential treatment for schizophrenia . Sanofi was originally investigating its potential use as a treatment for psychosis and anxiety . Following phase IIa clinical trials , osanetant entered phase IIb development in February 2001 . Osanetant was the first potent and selective non-peptide antagonist described for the NK3 tachykinin receptor . It has a higher affinity for human and guinea pig NK3 receptors than for rat NK3 receptors . In October 1999, Lehman Brothers predicted that the probability of the product reaching the market was 10%, with a possible launch in 2003 and potential peak sales of US $200 million in 2011 .
Sanofi-Aventis CEO, Chris Vihebacher,
PATENT
EP 0673928; FR 2717477; FR 2717478; FR 2719311; JP 1996048669; US 5741910; US 5942523; US 6124316
N-Benzyl-4-hydroxy-4-phenylpiperidine (II) was prepared by addition of phenyllithium to N-benzyl-4-piperidone (I). Carbinol (II) was then converted to acetamide (III) by acid-catalyzed Ritter reaction with acetonitrile. Replacement of the acetamido for an N-Boc group in (III) was effected by acidic hydrolysis of amide (III) to give (IV), followed by treatment with di-tert-butyl dicarbonate. The resultant 1-benzyl-4-(Boc-amino)-4-phenylpiperidine (V) was subjected to catalytic hydrogenolysis in the presence of Pd/C, and the N-debenzylated piperidine (VI) was reprotected as the N-trityl derivative (VII) by treatment with triphenylmethyl chloride and triethylamine. Reduction of the N-Boc group of (VII) by LiAlH4, yielded the N-methyl amine (VIII). After acylation of (VIII) with acetyl chloride to acetamide (IX), its N-trityl group was cleaved by treatment with hot aqueous formic acid to produce the intermediate piperidine (X).
![]()
![]()
Michael addition of methyl acrylate (XII) to (3,4-dichlorophenyl)acetonitrile (XI) produced the cyano diester adduct (XIII). Catalytic hydrogenation of the cyano group of (XIII) over Raney nickel with concomitant intramolecular cyclization gave rise to the piperidinone (XIV). After basic hydrolysis of the methyl ester function of (XIV), the resultant piperidone propionic acid (XV) was reduced to piperidino alcohol (XVI) by means of borane in THF. Resolution of the racemic piperidine (XVI) employing (+)-camphorsulfonic acid provided the dextro enantiomer (XVII). After N-protection of (XVII) as the Boc derivative (XVIII), its primary alcohol was activated as the corresponding mesylate (XIX) with methanesulfonyl chloride and Et3N. Condensation between mesylate (XIX) and intermediate piperidine (X) in acetonitrile at 60 C, produced (XX). The title benzamido derivative was then obtained by acid-promoted Boc group cleavage in (XX), followed by acylation with benzoyl chloride.
WO 9805640
Bioorg Med Chem Lett 1996,6(19),2307
In a related synthesis, (3,4-dichlorophenyl)acetonitrile (XI) was alkylated with bromide (XXII) –prepared by protection of 3-bromopropanol (XXI) with dihydropyran– to afford (XXIII). Subsequent Michael addition of methyl acrylate (XII) to (XXIII) in the presence of Triton B?gave the cyanoacid (XXIV). This was cyclized to the glutarimide (XXV) by refluxing in HOAc in the presence of H2SO4. Reduction of (XXV) using borane-dimethylsulfide complex produced the already reported racemic piperidinoalcohol (XVI). After acylation of the amine group of (XVI) with benzoyl chloride to yield (XXVI), its hydroxyl group was converted into the target mesylate precursor (XXVII) with methanesulfonyl chloride and Et3N.
![]()
An alternative preparation of the precursor 4-(N-methyl-N-acetyl)amino-4-phenylpiperidine (XXXIX) has been reported. The N-benzyl protecting group of piperidine (III) was replaced with an N-Boc group by catalytic hydrogenolysis to (XXXVI), followed by treatment with Boc2O to yield (XXXVII). Amide (XXXVII) alkylation with iodomethane under phase-transfer conditions gave the N-methyl derivative (XXXVIII). Subsequent N-Boc group cleavage in (XXXVIII) was accomplished by using zinc chloride in CH2Cl2 to afford the piperidine-ZnCl2 complex (XXXIX). This was then alkylated with mesylate (XXVII), and the title compound was finally isolated from the racemic mixture by means of preparative chiral HPLC.
![]()
![]()
In a further method, aminopiperidine (IV) was converted to the formamide (XL) by heating in ethyl formate. Formyl group reduction in (XL) with LiAlH4 provided the N-metyl amine (XLI). The N-benzyl group of (XLI) was then removed by catalytic hydrogenation over Pd/C. Alkylation of the resultant piperidine (XLII) with mesylate (XXVII) gave adduct (XLIII). After acetylation of (XLIII) in neat Ac2O, the racemic mixture was separated by chiral HPLC.
![]()
![]()
In a further procedure, nitrile (XXIII) was alkylated with ethyl 3-bromopropionate (XXVIII) to give cyano ester (XXIX). Catalytic hydrogenation of the cyano group of (XXIX) gave rise to the piperidinone (XXX), which was further reduced to piperidine (XXXI) with LiAlH4 in THF. Acid deprotection of the tetrahydropyranyl group of (XXXI), followed by resolution with (+)-camphorsulfonic acid, furnished the desired (S)-piperidinoalcohol camphorsulfonate salt (XXXII). Treatment of piperidine (XXXII) with benzoyl chloride in the presence of DIEA yielded benzamide (XXXIII). Conversion of the primary alcohol of (XXXIII) into the desired alkyl iodide (XXXV) was achieved via formation of the mesylate ester (XXXIV), followed by displacement of the mesylate group with KI in refluxing acetone.
Bioorg Med Chem Lett 1997,7(5),555
A new method has been reported. Formamide (XL) was prepared form carbinol (II) by a modified Ritter reaction with cyanotrimethylsilane. Subsequent reduction of (XL) with LiAlH4 gave the N-methyl amine (XLI), which was converted to acetamide (XLIV) by treatment with acetyl chloride. Benzyl group hydrogenolysis in (XLIV) afforded the piperidine (X). Finally, alkylation of piperidine (X) with the chiral alkyl iodide (XXXV) provided the title compound.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| US5741910 * |
Feb 29, 1996 |
Apr 21, 1998 |
Sanofi |
Compounds which are selective antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| US5942523 * |
Feb 29, 1996 |
Aug 24, 1999 |
Sanofi |
Compounds which are selective antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| US6040316 * |
Sep 2, 1997 |
Mar 21, 2000 |
Warner-Lambert Company |
3-alkyl-3-phenyl-piperidines |
| US6124316 * |
May 7, 1999 |
Sep 26, 2000 |
Sanofi |
Compounds which are specific antagonists of the human NK3 receptor and their use as medicinal products and diagnostic tools |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| US7648992 |
Jul 4, 2005 |
Jan 19, 2010 |
Astrazeneca Ab |
Hydantoin derivatives for the treatment of obstructive airway diseases |
| US7655664 |
Dec 14, 2005 |
Feb 2, 2010 |
Astrazeneca Ab |
Hydantoin derivatives as metalloproteinase inhibitors |
| US7662845 |
Aug 7, 2006 |
Feb 16, 2010 |
Astrazeneca Ab |
2,5-Dioxoimidazolidin-4-yl acetamides and analogues as inhibitors of metalloproteinase MMP12 |
| US7666892 |
May 5, 2008 |
Feb 23, 2010 |
Astrazeneca Ab |
Metalloproteinase inhibitors |
| US7700604 |
Dec 14, 2005 |
Apr 20, 2010 |
Astrazeneca Ab |
Hydantoin derivatives as metalloproteinase inhibitors |
| US7754750 |
|
Jul 13, 2010 |
Astrazeneca Ab |
Metalloproteinase inhibitors |
| US7989620 |
|
Aug 2, 2011 |
Astrazeneca Ab |
Hydantoin derivatives for the treatment of obstructive airway diseases |
| US8153673 |
Jan 26, 2010 |
Apr 10, 2012 |
Astrazeneca Ab |
Metalloproteinase inhibitors |
| US8183251 |
Nov 28, 2007 |
May 22, 2012 |
Astrazeneca Ab |
Hydantoin compounds and pharmaceutical compositions thereof |
| US20080032997 * |
Dec 14, 2005 |
Feb 7, 2008 |
Astrazeneca Ab |
Novel Hydantoin Derivatives as Metalloproteinase Inhibitors |
| US20080064710 * |
Jul 4, 2005 |
Mar 13, 2008 |
Astrazeneca Ab |
Novel Hydantoin Derivatives for the Treatment of Obstructive Airway Diseases |
| US20080221139 * |
Nov 28, 2007 |
Sep 11, 2008 |
David Chapman |
Novel Compounds |
| US20080262045 * |
May 5, 2008 |
Oct 23, 2008 |
Anders Eriksson |
Metalloproteinase Inhibitors |
| US20080293743 * |
Dec 14, 2005 |
Nov 27, 2008 |
Astrazeneca Ab |
Novel Hydantoin Derivatives as Metalloproteinase Inhibitors |
| US20080306065 * |
May 6, 2008 |
Dec 11, 2008 |
Anders Eriksson |
Metalloproteinase Inhibitors |
| US20100144771 * |
Dec 2, 2009 |
Jun 10, 2010 |
Balint Gabos |
Novel Hydantoin Derivatives for the Treatment of Obstructive Airway Diseases |
| WO2007106022A2 * |
Mar 15, 2007 |
Sep 20, 2007 |
Astrazeneca Ab |
A new crystalline form g of (5s) -5- [4- (5-chloro-pyridin-2- yloxy) -piperidine-1-sulfonylmethyl] – 5 -methyl -imidazolidine – 2,4-dione (i) and intermediates thereof. |
| WO2007106022A3 * |
Mar 15, 2007 |
Nov 1, 2007 |
Astrazeneca Ab |
A new crystalline form g of (5s) -5- [4- (5-chloro-pyridin-2- yloxy) -piperidine-1-sulfonylmethyl] – 5 -methyl -imidazolidine – 2,4-dione (i) and intermediates thereof. |
![]()
References
- “osanetant Sanofi-Aventis discontinued, France.”. Highbeam.
- Kamali, F (July 2001). “Osanetant Sanofi-Synthélabo”. Current opinion in investigational drugs (London, England : 2000). 2 (7): 950–6.PMID 11757797.
- Emonds-Alt, X; Bichon, D; Ducoux, JP; Heaulme, M; Miloux, B; Poncelet, M; Proietto, V; Van Broeck, D; et al. (1995). “SR 142801, the first potent non-peptide antagonist of the tachykinin NK3 receptor”. Life Sciences. 56 (1): PL27–32. doi:10.1016/0024-3205(94)00413-M.PMID 7830490.
- Quartara L, Altamura M (August 2006). “Tachykinin receptors antagonists: from research to clinic”. Current Drug Targets. 7 (8): 975–92.doi:10.2174/138945006778019381. PMID 16918326. Retrieved 2011-04-14.
- Desouzasilva, M; Mellojr, E; Muller, C; Jocham, G; Maior, R; Huston, J; Tomaz, C; Barros, M (May 2006). “The tachykinin NK3 receptor antagonist SR142801 blocks the behavioral effects of cocaine in marmoset monkeys”. European Journal of Pharmacology. 536 (3): 269–78.doi:10.1016/j.ejphar.2006.03.010. PMID 16603151.
- Jocham, Gerhard; Lezoch, Katharina; Müller, Christian P.; Kart-Teke, Emriye; Huston, Joseph P.; De Souza Silva, M. AngéLica (September 2006). “Neurokinin receptor antagonism attenuates cocaine’s behavioural activating effects yet potentiates its dopamine-enhancing action in the nucleus accumbens core”. European Journal of Neuroscience. 24 (6): 1721–32. doi:10.1111/j.1460-9568.2006.05041.x.PMID 17004936.
| X Emonds-Alt et al. SR 142801, the first potent non-peptide antagonist of the tachykinin NK3 receptor. Life Sci. 1995, 56(1), PL27-32. |
|
| F Kamali. Osanetant Sanofi-Synthélabo. Curr. Opin. Invest. Drugs. 2001, 2(7), 950-956. |
|
| L Quartara and M Altamura. Tachykinin receptors antagonists: from research to clinic. Curr. Drug Targets. 2006, 7(8), 975-992. |
|
| MA De Souza Silva et al. The tachykinin NK3 receptor antagonist SR142801 blocks the behavioral effects of cocaine in marmoset monkeys. Eur. J. Pharmacol. 2006, 536(3), 269-278. |
|
| G Jocham et al. Neurokinin receptor antagonism attenuates cocaine’s behavioural activating effects yet potentiates its dopamine-enhancing action in the nucleus accumbens core. Eur. J. Neurosci. 2006, 24(6), 1721-1732. |
///////Osanetant , SR-142,801,
CC(=O)N(C)C1(CCN(CC1)CCC[C@@]2(CCCN(C2)C(=O)C3=CC=CC=C3)C4=CC(=C(C=C4)Cl)Cl)C5=CC=CC=C5
![Day 13 of the 2016 Doodle Fruit Games! Find out more at g.co/fruit]()
Filed under:
Uncategorized Tagged:
801,
Osanetant,
SR-142 ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()