
Arformoterol
- MF C19H24N2O4
- MW 344.405
- Sunovion/Sepracor (Originator)
- Asthma Therapy, Bronchodilators, Chronic Obstructive Pulmonary Diseases (COPD), Treatment of, RESPIRATORY DRUGS, beta2-Adrenoceptor Agonists
- LAUNCHED 2007 , Phase III ASTHMA

Arformoterol is a long-acting β2 adrenoreceptor agonist (LABA) indicated for the treatment of chronic obstructive pulmonary disease(COPD). It is sold by Sunovion, under the trade name Brovana, as a solution of arformoterol tartrate to be administered twice daily (morning and evening) by nebulization.[1]
Arformoterol inhalation solution, a long-acting beta2-adrenoceptor agonist, was launched in the U.S. in 2007 for the long-term twice-daily (morning and evening) treatment of bronchospasm in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema. The product, known as Brovana(TM), for use by nebulization only, is the first long-acting beta2-agonist to be approved as an inhalation solution for use with a nebulizer. The product was developed and is being commercialized by Sunovion Pharmaceuticals (formerly Sepracor)
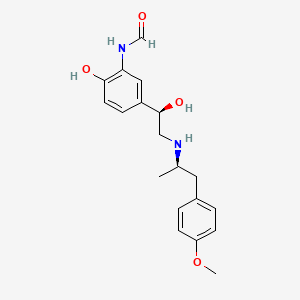
It is the active (R,R)-(−)-enantiomer of formoterol and was approved by the United States Food and Drug Administration (FDA) on October 6, 2006 for the treatment of COPD.
Arformoterol is a bronchodilator. It works by relaxing muscles in the airways to improve breathing. Arformoterol inhalation is used to prevent bronchoconstriction in people with chronic obstructive pulmonary disease, including chronic bronchitis and emphysema. The use of arformoterol is pending revision due to safety concerns in regards to an increased risk of severe exacerbation of asthma symptoms, leading to hospitalization as well as death in some patients using long acting beta agonists for the treatment of asthma.
Arformoterol is an ADRENERGIC BETA-2 RECEPTOR AGONIST with a prolonged duration of action. It is used to manage ASTHMA and in the treatment of CHRONIC OBSTRUCTIVE PULMONARY DISEASE.


Arformoterol tartrate
- Molecular FormulaC23H30N2O10
- Average mass494.492
- cas 200815-49-2
- 183-185°C
| N-[2-Hydroxy-5-[(1R)-1-hydroxy-2-[[(1R)-2-(4-methoxyphenyl)-1-methylethyl]amino]ethyl]phenyl]formamide (+)-(2R,3R)-Tartaric Acid; (-)-Formoterol 1,2-Dihydroxyethane-1,2-dicarboxylic Acid; (R,R)-Formoterol Threaric Acid; Arformoterol d-Tartaric Acid; Arformoterol d-α,β-Dihydroxysuccinic Acid |

SYNTHESIS
PATENT
us-9309186
Example 1
Synthesis of (R,R)-Formoterol-L-tartrate Form D
A solution containing 3.9 g (26 mmol) of L-tartaric acid and 36 mL of methanol was added to a solution of 9 g (26 mmol) of arformoterol base and 144 mL methanol at 23.degree. C. Afterwards, the resulting mixture was seeded with form D and stirred at 23.degree. C. for 1 hour. It was then further cooled to 0-5.degree. C. for 1 hour and the product collected by filtration and dried under inlet air (atmospheric pressure) for 16 hours to provide 11.1 g (86% yield) (99.7% chemical purity, containing 0.14% of the degradation impurity (R)-1-(3-amino-4-hydroxyphenyl)-2-[[(1R)-2-(4-methoxyphenyl)-1-methylethy- l]amino]ethanol) of (R,R)-formoterol L-tartrate form D, as an off white powder. .sup.1H-NMR (200 MHz, d.sub.6-DMSO) .delta.: 1.03 (d, 3H); 2.50-2.67 (m, 5H); 3.72 (s, 3H); 3.99 (s, 2H); 4.65-4.85 (m, 1H); 6.82-7.15 (m, 5H); 8.02 (s, 1H); 8.28 (s, 1H); 9.60 (s, NH). No residual solvent was detected (.sup.1H-NMR).
PSD: d.sub.50=2.3 .mu.m.


Taking Advantage of Polymorphism To Effect an Impurity Removal: Development of a Thermodynamic Crystal Form of (R,R)-FormoterolTartrate
Abstract

The development and large-scale implementation of a novel technology utilizing polymorphic interconversion and crystalline intermediate formation of (R,R)-formoterol l-tartrate ((R,R)-FmTA, 1) as a tool for the removal of impurities from the final product and generation of the most thermodynamically stable crystal form is reported. The crude product was generated by precipitation of the free base as the l-tartrate salt in a unique polymorphic form, form B. Warming the resultant slurry effected the formation of a partially hydrated stable crystalline intermediate, form C, with a concomitant decrease in the impurity levels in the solid. Isolation and recrystallization of form C provided 1 in the thermodynamically most stable polymorph, form A.





PATENT




Compound (R, R)-1-(4-Benzyloxy-3-nitro-phenyl)-2-[[2-(4-methoxy-phenyl)-1-methylethyl]-(1-phenyl-ethyl)-amino]-ethanol (compound VI), having the configuration represented by the following formula:

Examples(R)-2-(4-Benzyloxy-3-nitro-phenyl)-oxirane (II)
A solution of 90 g (0.25 mol) of (R)-1-(4-Benzyloxy-3-nitro-phenyl)-2-bromo-ethanol (compound I) in 320 mL of toluene and 50 mL of MeOH was added to a stirred suspension of 46 g (0.33 mol) of K2CO3 in 130 mL of toluene and 130 mL of MeOH. The mixture was stirred at 40°C for 20 h and washed with water (400 mL). The organic phase was concentrated under reduced pressure to a volume of 100 mL and stirred at 25 °C for 30 min. It was then further cooled to 0-5°C for 30 min. and the product collected by filtration and dried at 40 °C to provide 67.1 g (97% yield) (98% chemical purity, 100% e.e.) of compound II as an off-white solid. 1 H-NMR (200 MHz, CDCl3) δ: 2.80-2.90 (m, 2H); 3.11-3.20 (m, 2H), 3.80-3.90 (m, 1H); 5.23 (s, 2H); 7.11 (d, 2H); 7.41 (m, 5H), 7.76 (d, 2H).
Preparation of (R,R)-[2-(4-Methoxy-phenyl)-1-methyl-ethyl]-(1-phenyl-ethyl)-amine (III)
A solution of 13 g (78.6 mmol) of 1-(4-Methoxy-phenyl)-propan-2-one and 8.3 g (78.6 mmol) of (R)-1-Phenylethylamine in 60 mL MeOH was hydrogenated in the presence of 1.7 g of Pt/C 5% at 10 atm. and 30 °C for 20 h. The mixture was filtered though a pad of diatomaceous earth and concentrated under reduced pressure to give compound III as an oil. The obtained oil was dissolved in 175 mL of acetone, followed by addition of 6.7 mL (80.9 mmol) of a 12M HCl solution. The mixture was stirred at 23 °C for 30 min and at 0-5 °C for 30 min. The product collected by filtration and dried at 40 °C to provide 13.8 g of the hydrochloride derivate as a white solid. The obtained solid was stirred in 100 mL of acetone at 23 °C for 1h and at 0-5 °C for 30 min, collected by filtration and dried at 40 °C to provide 13.2 g of the hydrochloride derivate as a white solid. This compound was dissolved in 100 mL of water and 100 mL of toluene followed by addition of 54 mL (54 mmol) of 1N NaOH solution. The organic phase was concentrated to give 11.7 g (55% yield) (99% chemical purity and 100% e.e) of compound III as an oil.1H-NMR (200 MHz, CDCl3) δ: 0.88 (d, 3H); 1.31 (d, 3H), 2.40-2.50 (m, 1H); 2.60-2.80 (m, 2H); 3.74 (s, 3H); 3.90-4.10 (m, 1H); 6.77- 6.98 (m, 4H), 7.31 (s, 5H).
Synthesis of (R,R)-1-(4-Benzyloxy-3-nitro-phenyl)-2-[[2-(4-methoxy-phenyl)-1-methyl-ethyl]-(1-phenyl-ethyl)-amino]-ethanol (IV)
A 1-liter flask was charged with 50g (0.18 mol) of II and 50g (0.18 mol) of III and stirred under nitrogen atmosphere at 140 °C for 20 h. To the hot mixture was added 200 mL of toluene to obtain a solution, which was washed with 200 mL of 1N HCl and 200 mL of water. The organic phase was concentrated under reduced pressure to give 99 g (99% yield) (88% chemical purity) of compound IV as an oil. Enantiomeric purity 100%. 1H-NMR (200 MHz, CDCl3) δ: 0.98 (d, 3H); 1.41 (d, 3H), 2.60-2.90 (m, 4H); 3.20-3.30 (m, 1H); 3.74 (s, 3H); 4.10-4.20 (m, 1H); 4.30-4.40 (m, 1H), 5.19 (s, 2H); 6.69-7.42 (m, 16H); 7.77 (s, 1H).
Synthesis of (R, R)-1-(3-Amino-4-benzyloxy-phenyl)-2-[[2-(4-methoxy-phenyl)-1-methyl-ethyl]-(1-phenyl-ethyl)-amino]-ethanol (V)
A solution of 99 g (0.18 mol) of IV in 270 mL IPA and 270 mL toluene was hydrogenated in the presence of 10 g of Ni-Raney at 18 atm and 40 °C for 20 h. The mixture was filtered though a pad of diatomaceous earth and the filtrate was concentrated under reduced pressure to give 87 g (92% yield) (83% chemical purity, 100 % e.e.) of compound V as an oil. 1H-NMR (200 MHz, CDCl3) δ: 0.97 (d, 3H); 1.44 (d, 3H), 2.60-2.90 (m, 4H); 3.20-3.30 (m, 1H); 3.74 (s, 3H); 4.10-4.20 (m, 1H); 4.30-4.40 (m, 1H), 5.07 (s, 2H); 6.67-6.84 (m, 7H); 7.25-7.42 (m, 10H).
Synthesis of (R,R)-N-(2-Benzyloxy-5-{1-hydroxy-2-[[2-(4-methoxy-phenyl)-1-methyl-ethyl]-(1-phenyl-ethyl)-amino]-ethyl)-phenyl)-formamide (VI)
24 mL (0.63 mol) of formic acid was added to 27 mL (0.28 mol) of acetic anhydride and stirred at 50 °C for 2 h under nitrogen atmosphere. The resulting mixture was diluted with 100 mL of CH2Cl2 and cooled to 0 °C. A solution of 78 g (0.15 mol) of V in 300 mL de CH2Cl2 was slowly added and stirred for 1h at 0 °C. Then, 150 mL of 10% K2CO3 aqueous solution were added and stirred at 0 °C for 15 min. The organic phase was washed twice with 400 mL of 10% K2CO3 aqueous solution and concentrated under reduced pressure to give 80 g (97% yield, 100% e.e.) (75% chemical purity) of compound VI as an oil. 1H-NMR (200 MHz, CDCl3) δ: 0.98 (d, 3H); 1.42 (d, 3H), 2.60-2.90 (m, 4H); 3.20-3.30 (m, 1H); 3.75 (s, 3H); 4.10-4.20 (m, 1H); 4.30-4.40 (m, 1H), 5.09 (s, 2H); 6.67-7.41 (m, 17H); 8.4 (d, 1H).
Synthesis (R,R)-N-(2-Hydroxy-5-{1-hydroxy-2-[2-(4-methoxy-phenyl)-1-methyl-ethylamino]-ethyl}-phenyl)-formamide (VII)
A solution of 8.5 g (16 mmol) of VI, previous purified by column chromatography on silica gel (AcOEt/heptane, 2:3), in 60 mL ethanol was hydrogenated in the presence of 0.14 g of Pd/C 5% at 10 atm. and 40 °C for 20 h. The mixture was filtered though a pad of diatomaceous earth and concentrated under reduced pressure to give 5 g (93% yield) (91% chemical purity, 100% e.e.) of compound VII as foam. m. p.= 58-60 °C. 1H-NMR (200 MHz, d6-DMSO) δ: 0.98 (d, 3H); 2.42-2.65 (m, 5H); 3.20-3.40 (m, 1H); 3.71 (s, 3H); 4.43-4.45 (m, 1H); 6.77-7.05 (m, 5H); 8.02 (s, 1H), 8.26 (s, 1H).
Synthesis (R,R)-N-(2-Hydroxy-5-{1-hydroxy-2-[2-(4-methoxy-phenyl)-1-methyl-ethylamino]-ethyl}-phenyl)-formamide (VII)
A solution of 46 g (0.08 mol) of VI, crude product, was dissolved in 460 mL ethanol and hydrogenated in the presence of 0.74 g of Pd/C 5% at 10 atm. and 40 ° C for 28 h. The mixture was filtered though a pad of diatomaceous earth and the filtrate was concentrated under reduced pressure to give 24 g (83% yield) (77% chemical purity, 100% e.e.) of compound VII as a foam. m. p. = 58-60 °C. 1H-NMR (200 MHz, d6-DMSO) δ: 0.98 (d, 3H); 2.42-2.65 (m, 5H); 3.20-3.40 (m, 1H); 3.71 (s, 3H); 4.43-4.45 (m, 1H); 6.77-7.05 (m, 5H); 8.02 (s, 1H), 8.26 (s, 1H).
The HPLC conditions used for the determination of the Chemical purity % are described in the table below:
-
HPLC Column Kromasil 100 C-18 Dimensions 0.15 m x 4.6 mm x 5 µm Buffer 2.8 ml TEA (triethylamine) pH=3.00 H3PO4 (85%) in 1 L of H2O Phase B Acetonitrile Flow rate 1.5 ml miN-1 Temperature 40 °C Wavelength 230 nm The HPLC conditions used for the determination of the enantiomeric purity % are described in the table below:
HPLC Column Chiralpak AD-H Dimensions 0.25 m x 4.6 mm Buffer n-hexane : IPA : DEA (diethyl amine) : H2O 85:15:0.1:0.1 Flow rate 0.8 ml min-1 Temperature 25 °C Wavelength 228 nm
PATENT
Example 1
(R) -2- (4- benzyloxy-3-nitrophenyl) oxirane (I) (9. 86g, 36mmol) and (R) -I- (4- methoxy- phenyl) -N – [(R) -I- phenyl-ethyl] -2-amino-propane (II) (10. 8g, 40mmol) cast in the reaction flask, the reaction 20 hours at 140 ° C, the chiral Intermediate (III) (17. 3g, yield 88%). HPLC: de values of> 90%; MS (ESI) m / z: 541 3 (M ++ 1); 1H-NMR (CDCl3):.. Δ 0. 96 (d, 3H), 1 49 (d, 3H ), 2 · 15 (q, 1Η), 2 · 67 (dq, 2H), 2. 99 (dq, 2H), 3. 74 (s, 3H), 4. 09 (d, 1H), 4. 56 (q, 1H), 5. 24 (s, 2H), 6. 77 (dd, 4H), 7. 10 (d, 1H), 7. 25-7. 5 (m, 11H), 7. 84 ( s, 1H).
Example 2
(R) -2- (4- benzyloxy-3-nitrophenyl) oxirane (I) (9. 86g, 36mmol) and (R) -I- (4- methoxybenzene yl) -N – [(R) -I- phenyl-ethyl] -2-amino-propane (II) (10. 8g, 40mmol) and toluene 100ml, 110 ° C0-flow reactor 36 hours, the solvent was distilled off succeeded intermediates (III) (16. 8g, yield 85%).
Example 3
(R) -2- (4- benzyloxy-3-nitrophenyl) oxirane (I) (9. 86g, 36mmol) and (R) -I- (4- methoxybenzene After [(R) -I- phenyl-ethyl] -2-amino-propane (II) (10. 8g, 40mmol) and dichloromethane 100ml, 30 ° C for 48 hours, and the solvent was distilled off – yl) -N succeeded intermediates (III) (15. Sg, yield 80%).
Example 4
(R) -2- (4- benzyloxy-3-nitrophenyl) oxirane (I) (9. 86g, 36mmol) and (R) -I- (4- methoxybenzene yl) -N – [(R) -I- phenyl-ethyl] -2-amino-propane (II) (8. 75g, 32mmol) cast in the reaction flask, the reaction 20 hours at 140 ° C, the chiral intermediate form (III) (16. 3g, 83% yield).
Example 5
(R) -2- (4- benzyloxy-3-nitrophenyl) oxirane (I) (9. 86g, 36mmol) and (R) -I- (4- methoxybenzene yl) -N – [(R) -I- phenyl-ethyl] -2-amino-propane (II) (14. 6g, 54mmol) cast in the reaction flask, the reaction 20 hours at 140 ° C, the chiral intermediate form (III) (17. 5g, 89% yield).

Scheme
product (SS) W semifumarate (109 mg) characterized by ‘HNMR (4-D MSO) 6 (ppm) 1.00 (d, 3H, CHCH,), 4.624.70 (m, lH,
CHOH), 3.73 (s, 3H, OCH,), 6.M.9 (m, 3H, aromatic), 7.00 (dd,4H, aromatic), 6.49 (s, 1@ CH = CH fumarate). MS of disilylated
(SS) W: 473 (M +<H3,7%); 367 (M ‘<8H90, 45%); 310 61%). The (RSS) fraction was treated in the same manner
giving the product (R;S) W semifumarate, which was characterized by ‘H-NMR (4-DMSO) 6 (ppm) 1.01 (d, 3H, CHCH,),
3.76 (s, 3H, OC&), 6.49 (s, lH, CH=CH, fumarate) 6.M.9 (m, 3H, aromatic), 7.0 (dd, 4H, aromatic). MS of disilylated (R;S)
(M’X~~HIGNO1,7 %); 178 ( C I ~H~ ~N95O%,) ; 121 (CsH90, W. 473 (M’4H3, 5%); 367 (M’4gH90, 48%); 310
(M +–CI~HIGNO18, %); 178 (CIIHIGNO, 95%); 121 (CsH90, 52%). The structural data for the (RR) and (S;R) enantiomers
were in accordance with the proposed structures. The enantiomeric purity obtained for the enantiomers in each batch is
shown in Table 1.


Large-Scale Synthesis of Enantio- and Diastereomerically Pure (R,R)-Formoterol†
Abstract
(R,R)-Formoterol (1) is a long-acting, very potent β2-agonist, which is used as a bronchodilator in the therapy of asthma and chronic bronchitis. Highly convergent synthesis of enantio- and diastereomerically pure (R,R)-formoterol fumarate is achieved by a chromatography-free process with an overall yield of 44%. Asymmetric catalytic reduction of bromoketone 4 using as catalyst oxazaborolidine derived from (1R, 2S)-1-amino-2-indanol and resolution of chiral amine 3 are the origins of chirality in this process. Further enrichment of enantio- and diastereomeric purity is accomplished by crystallizations of the isolated intermediates throughout the process to give (R,R)-formoterol (1) as the pure stereoisomer (ee, de >99.5%).


Scheme


Formoterol is a long-acting β2-adrenoceptor agonist and has a long duration of action of up to 12 hours. Chemically it is termed as Λ/-[2-hydroxy-5-[1-hydroxy-2-[[2-(4- methoxyphenyl)propan-2-yl]amino]ethyl]phenyl]-formamide. The structure of formoterol is as shown below.

The asterisks indicate that formoterol has two chiral centers in the molecule, each of which can exist in two possible configurations. This gives rise to four diastereomers which have the following configurations: (R,R), (S1S), (S1R) and (R1S).
(R1R) and (S1S) are mirror images of each other and are therefore enantiomers. Similarly (S1R) and (R1S) form other enatiomeric pair.
The commercially-available formoterol is a 50:50 mixture of the (R1R)- and (S1S)- enantiomers. (R,R)-formoterol is an extremely potent full agonist at the β2-adrenoceptor and is responsible for bronchodilation and has anti-inflammatory properties. On the other hand (S,S)-enantiomer, has no bronchodilatory activity and is proinflammatory.
Murase et al. [Chem.Pharm.Bull., .26(4)1123-1129(1978)] synthesized all four isomers of formoterol and examined for β-stimulant activity. In the process, racemic formoterol was subjected to optical resolution with tartaric acid.
In another attempt by Trofast et al. [Chirality, 3:443-450(1991 )], racemic 4-benzyloxy-3- nitrostryrene oxide was coupled with optically pure N-[(R)-1-phenylethyl]-2-(4- methoxyphenyl)-(R)1-methylethylamine to give diastereomeric mixtures of intermediates, which were separated by column chromatography and converted to the optically pure formoterol.
In yet another attempt, racemic formoterol was subjected to separation by using a chiral compound [International publication WO 1995/018094].
WO 98/21175 discloses a process for preparing optically pure formoterol using optically pure intermediates (R)-N-benzyl-2-(4-methoxyphenyl)-1-methylethyl amine and (R)-4- benzyloxy-3-formamidostyrene oxide.
Preparation of optically pure formoterol is also disclosed in IE 000138 and GB2380996.

Example 7
Preparation of Arformoterol
4-benzyloxy-3-formylamino-α-[N-benzyl-N-(1-methyl-2-p- methoxyphenylethyl)aminomethyl]benzyl alcohol (120gms, 0.23M), 10% Pd/C (12 gms) and denatured spirit (0.6 lit) were introduced in an autoclave. The reaction mass was hydrogenated by applying 4 kg hydrogen pressure at 25-300C for 3 hrs. The catalyst was removed by filtration and the, clear filtrate concentrated under reduced pressure below 400C to yield the title compound. (63 gms, 80%).
Example 8
Preparation of Arformoterol Tartrate
Arformoterol base (60 gms, 0.17M), 480 ml IPA , 120 ml toluene and a solution of l_(+)- tartaric acid (25.6 gms, 0.17M) in 60 ml distilled water were stirred at 25-300C for 2 hrs and further at 40°- 45°C for 3 hrs. The reaction mass was cooled to 25-300C and further chilled to 200C for 30 mins. The solid obtained was isolated by filtration to yield the title compound. (60 gms, 70%),
The tartrate salt was dissolved in hot 50% IPA-water (0.3 lit), cooled as before and filtered to provide arformoterol tartrate. (30 gms, 50 % w/w). having enantiomeric purity greater than 99%.
Modulation of Catalyst Reactivity for the Chemoselective Hydrogenation of a Functionalized Nitroarene: Preparation of a Key Intermediate in the Synthesis of (R,R)-Formoterol Tartrate

PAPER
Francisco Campos, M. Pilar Bosch and Angel Guerrero*





The solution was left standing overnight and the resulting crystalline solid (7.6 mg) puri®ed on areverse-phase column (1 g, Isolute SPE C18) using mixtures of MeOH±H2O as eluent. The solventwas removed under vacuum and the aqueous solution lyophilized (^35C, 0.6 bar) overnight. The(l)-(+)-tartrate salt of (R,R)-1 showed an 20D=-29.4 (H2O, c 0.61) (>99% ee based on the
reported value 34). 34=Hett, R.; Senanayake, C. H.; Wald, S. A. Tetrahedron Lett. 1998, 39, 1705.
Diethylanilineborane: A Practical, Safe, and Consistent-Quality Borane Source for the Large-Scale Enantioselective Reduction of a Ketone Intermediate in the Synthesis of (R,R)-Formoterol
Abstract

The development of a process for the use of N,N-diethylaniline−borane (DEANB) as a borane source for the enantioselective preparation of a key intermediate in the synthesis of (R,R)-formoterol l-tartrate, bromohydrin 2, from ketone 3 on kilogram scale is described. DEANB was found to be a more practical, safer, and higher-quality reagent when compared to other more conventional borane sources: borane−THF and borane−DMS.
PAPER
http://nopr.niscair.res.in/bitstream/123456789/8917/1/IJCB%2044B(1)%20167-169.pdf


PAPER
http://www.bioorg.org/down/Hetetorcycles_07_2243.pdf?ckattempt=1



PAPER
Arformoterol: (R,R)-eformoterol, (R,R)-formoterol, arformoterol tartrate, eformoterol-sepracor, formoterol-sepracor, R,R-eformoterol, R,R-formoterol.
Abstract
Sepracor in the US is developing arformoterol [R,R-formoterol], a single isomer form of the beta(2)-adrenoceptor agonist formoterol [eformoterol]. This isomer contains two chiral centres and is being developed as an inhaled preparation for the treatment of respiratory disorders. Sepracor believes that arformoterol has the potential to be a once-daily therapy with a rapid onset of action and a duration of effect exceeding 12 hours. In 1995, Sepracor acquired New England Pharmaceuticals, a manufacturer of metered-dose and dry powder inhalers, for the purpose of preparing formulations of levosalbutamol and arformoterol. Phase II dose-ranging clinical studies of arformoterol as a longer-acting, complementary bronchodilator were completed successfully in the fourth quarter of 2000. Phase III trials of arformoterol began in September 2001. The indications for the drug appeared to be asthma and chronic obstructive pulmonary disease (COPD). However, an update of the pharmaceutical product information on the Sepracor website in September 2003 listed COPD maintenance therapy as the only indication for arformoterol. In October 2002, Sepracor stated that two pivotal phase III studies were ongoing in 1600 patients. Sepracor estimates that its NDA submission for arformoterol, which is projected for the first half of 2004, will include approximately 3000 adult subjects. Sepracor stated in July 2003 that it had completed more than 100 preclinical studies and initiated or completed 15 clinical studies for arformoterol inhalation solution for the treatment of bronchospasm in patients with COPD. In addition, Sepracor stated that the two pivotal phase III studies in 1600 patients were still progressing. In 1995, European patents were granted to Sepracor for the use of arformoterol in the treatment of asthma, and the US patent application was pending.
CLIP



PAPER
doi:10.1016/j.cclet.2008.01.012
http://www.sciencedirect.com/science/article/pii/S1001841708000132
Volume 19, Issue 3, March 2008, Pages 279–280

New method in synthesizing an optical active intermediate for (R,R)-formoterol
- Key Laboratory of Drug Targeting Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China\
Abstract
(R)-1-(4-Methoxyphenyl)propan-2-amine 2a, an optical active intermediate for (R,R)-formoterol, was synthesized from d-alanine in 65% overall yield by using a simple route, which contained protecting amino group, cyclization, coupling with Grignard reagent, reduction and deprotection.


References
| Muller, P., et al.: Arzneimittel-Forsch., 33, 1685 (1983); Wallmark, B., et al.: Biochim. Biophys. Acta., 778, 549 (1984); Morii, M., et al.: J. Biol. chem., 268, 21553 (1993); Ritter, M., et al.: Br. J. Pharmacol., 124, 627 (1998); Stenhoff, H., et al.: J. Chromatogr., 734, 191 (1999), Johnson, D.A., et al.: Expert Opin. Pharmacother., 4, 253 (2003); Bouyssou, T., et al.: Bio. Med. Chem. Lett. 20, 1410, (2010); |
- “Brovana Prescribing information, Dosage and Administration section”. Archived from the original on 13 February 2008. Retrieved2008-03-14.
- SEE http://pubs.acs.org/doi/abs/10.1021/jm5013227
External links
- Brovana website
| EP0390762A1 * | 23 Mar 1990 | 3 Oct 1990 | Aktiebolaget Draco | New bronchospasmolytic compounds and process for their preparation |
| EP0938467A1 | 7 Nov 1997 | 1 Sep 1999 | Sepracor, Inc. | Process for the preparation of optically pure isomers of formoterol |
| EP1082293A2 | 20 May 1999 | 14 Mar 2001 | Sepracor Inc. | Formoterol polymorphs |
| WO2009147383A1 | 2 Jun 2009 | 10 Dec 2009 | Cipla Limited | Process for the synthesis of arformoterol |
| Reference | ||
|---|---|---|
| 1 | * | HETT R ET AL: “Enantio- and Diastereoselective Synthesis of all Four Stereoisomers of Formoterol” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL LNKD- DOI:10.1016/S0040-4039(97)00088-9, vol. 38, no. 7, 17 February 1997 (1997-02-17), pages 1125-1128, XP004034214 ISSN: 0040-4039 |
| 2 | * | LING HUANG ET AL.: “The Asymmetric Synthesis of (R,R)-Formoterol via Transfer Hydrogenation with Polyethylene Glycol Bound Rh Catalyst in PEG2000 and Water” CHIRALITY, vol. 22, 30 April 2009 (2009-04-30), pages 206-211, XP002592699 |
| 3 | MURASE ET AL. CHEM. PHARM. BULL. vol. 26, no. 4, 1978, pages 1123 – 1129 | |
| 4 | TROFAST ET AL. CHIRALITY vol. 1, 1991, page 443 | |
| 5 | * | TROFAST J ET AL: “STERIC ASPECTS OF AGONISM AND ANTAGONISM AT BETA-ADRENICEPTORS: SYNTHESIS OF AND PHARMACOLOGICAL EXPERIMENTS WITH THE ENANTIOMERS OF FORMOTEROL AND THEIR DIASTEREOMERS” CHIRALITY, WILEY-LISS, NEW YORK, US LNKD- DOI:10.1002/CHIR.530030606, vol. 3, no. 6, 1 January 1991 (1991-01-01) , pages 443-450, XP002057060 ISSN: 0899-0042 |
| 6 | WILKINSON, H.S ET AL. ORGANIC PROCESS RESEARCH AND DEVELOPMENT vol. 6, 2002, pages 146 – 148 | |
Durham E-Theses A Solid-state NMR Study of Formoterol Fumarate
|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[2-hydroxy-5-[(1R)-1-hydroxy-2-[[(2R)-1-(4-methoxyphenyl) propan-2-yl]amino]ethyl] phenyl]formamide
|
|
| Clinical data | |
| Trade names | Brovana |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602023 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Inhalation solution fornebuliser |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 52–65% |
| Biological half-life | 26 hours |
| Identifiers | |
| CAS Number | 67346-49-0  |
| ATC code | none |
| PubChem | CID 3083544 |
| IUPHAR/BPS | 7479 |
| DrugBank | DB01274 |
| ChemSpider | 2340731 |
| UNII | F91H02EBWT |
| ChEBI | CHEBI:408174 |
| ChEMBL | CHEMBL1201137  |
| Chemical data | |
| Formula | C19H24N2O4 |
| Molar mass | 344.405 g/mol |

Formoterol
//////Arformoterol, (R,R)-Formoterol, (R,R)-Formoterol-L-(+)-tartrate, 200815-49-2, Arformoterol tartrate , Brovana, UNII:5P8VJ2I235, Sepracor, Asthma Therapy, Bronchodilators, Chronic Obstructive Pulmonary Diseases, COPD , RESPIRATORY DRUGS, beta2-Adrenoceptor Agonists, Phase III, 2007, Sunovion
COC1=CC=C(C[C@@H](C)NC[C@H](O)C2=CC(NC=O)=C(O)C=C2)C=C1
Filed under: GENERICS Tagged: (R)-, 2007, 200815-49-2, Arformoterol, Arformoterol tartrate, Asthma Therapy, beta2-Adrenoceptor Agonists, Bronchodilators, Brovana, Chronic Obstructive Pulmonary Diseases, COPD, Phase III, R)-Formoterol, R)-Formoterol-L-(+)-tartrate, RESPIRATORY DRUGS, Sepracor, Sunovion, UNII:5P8VJ2I235







