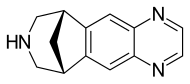
Varenicline (Chantix™)
Varenicline
- MF C13H13N3
- MW 211.26
Varenicline (trade name Chantix and Champix usually in the form of varenicline tartrate), is a prescription medication used to treatnicotine addiction. Varenicline is a nicotinic receptor partial agonist—it stimulates nicotine receptors more weakly than nicotine itself does. In this respect it is similar to cytisine and different from the nicotinic antagonist, bupropion, and nicotine replacement therapies(NRTs) like nicotine patches and nicotine gum. As a partial agonist it both reduces cravings for and decreases the pleasurable effects of cigarettes and other tobacco products. Through these mechanisms it can assist some patients to quit smoking.



Varenicline Tartrate

C13H13N3
 C4H6O6 : 361.35
C4H6O6 : 361.35[375815-87-5]
Medical uses
Varenicline is used for smoking cessation. In a 2009 meta-analysis varenicline was found to be more effective than bupropion (odds ratio 1.40) and NRTs (odds ratio 1.56).[1]
A 2013 Cochrane overview and network meta-analysis concluded that varenicline is the most effective medication for tobacco cessation and that smokers were nearly three times more likely to quit on varenicline than with placebo treatment. Varenicline was more efficacious than bupropion or NRT and as effective as combination NRT for tobacco smoking cessation.[2][3]
The United States’ Food and Drug Administration (US FDA) has approved the use of varenicline for up to twelve weeks. If smoking cessation has been achieved it may be continued for another twelve weeks.[4]
Varenicline has not been tested in those under 18 years old or pregnant women and therefore is not recommended for use by these groups. Varenicline is considered a class C pregnancy drug, as animal studies have shown no increased risk of congenital anomalies, however, no data from human studies is available.[5] An observational study is currently being conducted assessing for malformations related to varenicline exposure, but has no results yet.[6] An alternate drug is preferred for smoking cessation during breastfeeding due to lack of information and based on the animal studies on nicotine.[7]

Varenicline L-tartrate (Compound I) is the international commonly accepted name for 7,8,9,10- tetrahydro-6, 10-methano-6i7-pyrazino [2, 3- h] [3 ] benzazepme, (2R, 3R) -2 , 3-dihydroxybutanedioate (1:1) (which is also known as 5,8,14- tπazatetracyclo [10.3.1. O2‘11. O4‘9] -hexadeca-2 (11) , 3, 5, 7, 9-pentaene, (2R, 3R)-2,3- dihydroxybutanedioate (1:1)) and has an empirical formula of C13H13N3 • C4H6O6 and a molecular weight of 361.35. Varenicline L-tartrate is a commercially marketed pharmaceutically active substance known to be useful for the treatment of smoking addiction.

(D
Varenicline L-tartrate is a partial agonist selective for (X4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the name Chantix™ for the treatment of smoking cessation. Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550. In particular, Example 26 of U.S. Patent No. 6,410,550 describes the preparation of varenicline hydrochloride salt using 1- (4 , 5-dinitro-10- aza-tπcyclo [6.3.1.O2‘7] dodeca-2, 4, 6-trien-10-yl) -2,2,2- tπfluoroethanone (compound of formula (III)) as starting compound. On the other hand, Example HA) of U.S. Patent No. 6,410,550 illustrates the preparation of compound of formula (III) via nitration of compound of formula (II) using an excess of nitronium triflate (>4 equiv) as a nitrating agent. The process disclosed in U.S. Patent No. 6,410,550 is depicted in Scheme 1.

VareniclineΗCl
Scheme 1
However, Coe et al., J. Med. Chem., 48, 3474 (2005), describes the same process and examples as U.S. Patent No. 6,410,550, and it also reveals that this process affords intermediate ortho-4 , 5-dinitrocompound of formula (III) together with the meta-3, 5-dinitro- isomer (i.e. the meta-dinitrocompound) in a ratio 9:1. The presence of the meta-dinitrocompound may affect not only the purity of the intermediate compound of formula III but it may also have an effect on the purity of the final varenicline tartrate, given that it can be carried along the synthetic pathway and/or it can also give rise to other derivative impurities. Thereby, as well as in U.S. Patent No. 6,410,550, in order to isolate pure compound of formula (III) , the raw product is triturated with ethyl acetate/hexane to afford compound of formula (III) with 77% yield. Additionally, the mother liquor is purified by chromatography on silica gel to improve the yield to a total of 82.8%. However, this process is not desirable for industrial implementation since it requires extensive and complicated purification procedures, i.e. trituration of the solid product along with column chromatography purification of the mother liquor, which is not very efficient or suitable for industrial scale-up.
Several improved processes for the synthesis of varenicline or its salts have been reported in the literature (e.g. WO2006/090236) . However, none of these processes tackle the optimization of the purification step of compound of formula (III).
There is therefore the need for providing an improved process for the preparation of varenicline L- tartrate which involves simple experimental procedures well suited to industrial production, which avoids the use of column chromatography purifications, and which affords high pure varenicline L-tartrate which hence can be used directly as a starting product for the preparation of the marketed pharmaceutical speciality.
Additionally, it has been observed that varenicline L-tartrate is usually obtained as a yellow solid under – A –
standard synthetic conditions. In this regard, colour must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC. The presence of impurities may adversely affect the safety and shelf life of formulations. In this connection, International application No. WO2006/090236 describes the isolation of vareniclme L- tartrate as a white solid. However, in order to remove coloured impurities, the varenicline L-tartrate obtained in WO2006/090236 is treated with a particular activated carbon having a specific grade (i.e. Darco KB-B™) . In fact, Example 5 of WO2006/090236 describes a large reprocessing step which comprises: dissolving varenicline L-tartrate in water, adding toluene, basifying with NaOH aqueous solution, collecting the toluene phase containing varenicline free base, distilling, adding methanol, azeotropically distilling the mixture, and adding more methanol to obtain a methanolic solution containing varenicline free base, adding Darco KB-B™ (10% w/w) , stirring for one hour, filtering through a pad of celite, and treating with L-tartaric acid to give varenicline L- tartrate salt as a white solid. Further, WO2006/090236 provides the absorbance at 430 nm of a varenicline L- tartrate salt solution, either in dichloromethane or in toluene, with or without using Darco KB-B™ activated carbon. However, this measure cannot be used to corroborate the whiteness of the solid varenicline L- tartrate. In addition, Example 3 of International application No. WO2002/092089, also disclose the preparation of varenicline L-tartrate polymorphic form C (i.e. a hydrate polymorph) as a white precipitate. Therefore, there is also a need for a simple and efficient method for preparing varenicline L-tartrate with enhanced whiteness and having a high purity.
SYNTHESIS



Synthesis of Intermediate VIII
Paper
J. Med. Chem. 48, 3474 (2005).
http://pubs.acs.org/doi/pdf/10.1021/jm050069n

PATENT
https://www.google.com/patents/WO2001062736A1?cl=en
CLIP
Profiles of Drug Substances, Excipients and Related Methodology, Volume 37
edited by Harry G. Brittain







SYNTHESIS

|
DOI: 10.1021/jm00190a020
|
|
| DOI: 10.1021/jm050069n |
CLIP
Scheme (I) compound patent US6410550B1 is provided adjacent difluorobromobenzene as raw materials by DA reaction, oxidation, cyclization, debenzylation get varenicline intermediate (II). The synthesis route is as follows:

Patent CN101693712A mainly given varenicline intermediate (II) The preparation process is different from the compound patented. After the five-step method patents cited compounds. The entire route is longer, while using a large number of precious metal catalysts and reaction conditions need very strict control, inappropriate EVAL industry production.



PATENT
A varenicline intermediate 2,3, 4, 5-tetrahydro-1,5-methylene bridge synthesis -1H-3- benzazepine hydrochloride, which comprises the following Step: (1) 2-indanone of formula 3 and the compound and paraformaldehyde under alkaline or acidic conditions Mannich reaction, as shown in general formula 2 intermediate; (2) the step (I) obtained through reaction of Formula 2 intermediate under basic or acidic conditions by reducing the role of the carbonyl group is reduced to a methylene group, and get varenicline intermediate (II) by debenzylation, the reaction is:

Wherein, R groups are selected from _H, _Me, _Et, _iPr> _t_Bu.
Figure 2;

Wherein, R group is -H, -Me, -Et, -iPr or -t_Bu.
(2) Step (I) obtained by the reaction intermediates of formula under basic or acidic conditions by reducing the role of the carbonyl group is reduced 2 methylene, and get by debenzylation cutting Lenk Lin intermediate (II);


CLIP
Varenicline, a nicotinic 42 partial agonist, was approved in the US for the treatment of smoking cessation in May of 2006. It was developed and marketed by Pfizer as a treatment for cigarette smokers who want to quit. Varenicline partially activates the nicotinic receptors and thus reduces the craving for cigarette that smokers feel when they try to quit smoking. By mitigating this craving and antagonizing nicotine activity without other symptoms, this novel drug helps quitting this dangerous addiction easier on the patients [6,52]. Several modifications [54,55] to the original synthesis [53,56] have been reported in the literature, including an improved process scale synthesis of the last few steps (Scheme 15) [57]. The Grignard reaction was initiated on a small scale by addition of 2-bromo fluorobenzene 113 to a slurry of Magnesium turnings and catalytic 1,2-dibromoethane in THF and heating the mixture until refluxing in maintained. To this refluxing mixture was added a mixture of the 2-bromo fluorobenzene 113 and cyclopentadiene 114 over a period of 1.5 h. After complete addition, the reaction was allowed to reflux for additional 1.5 h to give the Diels- Alder product 115 in 64% yield. Dihydroxylation of the olefin 115 by reacting with catalytic osmium tetraoxide in the presence of N-methylmorpholine N-oxide (NMO) in acetone: water mixture at room temperature provided the diol 116 in 89% yield. Oxidative cleavage of diol 116 with sodium periodate in biphasic mixture of water: DCE at 10ºC provided di-aldehyde 117 which was immediately reacted with benzyl amine in the presence of sodium acetoxyborohydride to give benzyl amine 118 in 85.7% yield. The removal of the benzyl group was effected by hydrogenation of the HCl salt in 40-50 psi hydrogen pressure with 20% Pd(OH)2 in methanol to give amine hydrochloride 119 in 88% yield. Treatment of amine 119 with trifluoroacetic anhydride and pyridine in dichloromethane at 0ºC gave trifluoroacetamide 120 in 94% yield. Dinitro compound 121 was prepared by addition of trifluoroacetamide 120 to a mixture of trifluoromethane sulfonic acid and nitric acid, which was premixed, in dichloromethane at 0ºC. Reduction of the dinitro compound 121 by hydrogenation at 40-50 psi hydrogen in the presence of catalytic 5%Pd/C in isopropanol:water mixture provided the diamine intermediate 122 which was quickly reacted with glyoxal in water at room temperature for 18h to give compound 123 in 85% overall yield. The trifluoroacetamide 123 was then hydrolyzed with 2 M sodium hydroxide in toluene at 37-40ºC for 2-3h followed by preparation of tartrate salt in methanol to furnish varenicline tartrate (XV).

[52]Keating, G.; Siddiqui, M. A. A. CNSdrugs, 2006, 11, 946.
[53] Coe, J. W.; Brooks, P. R.; Vetelino, M. G.; Wirtz, M. C.; Arnold,E. P. ; Huang, J.; Sands, S. B.; Davis, T. I.; Lebel, L. A.; Fox, C.
B.; Shrikhande, A.; Heym, J. H.; Schaeffer, E.; Rollema, H.; Lu,Y.; Mansbach, R. S.; Chambers, L. K.; Rovetti, C. C.; Schulz, D.
W.; Tingley, III, F. D.; O’Neill, B. T. J. Med. Chem., 2005, 48,3474.
[54] Brooks, P. R.; Caron, S.; Coe, J. W.; Ng, K. K.; Singer, R. A.;Vazquez, E.; Vetelino, M. G.; Watson, Jr. H. H.; Whritenour, D.
C.; Wirtz, M. C. Synthesis, 2004, 11, 1755.
[55] Singer, R. A.; McKinley, J. D.; Barbe, G.; Farlow, R. A. Org. Lett.,2004, 6, 2357.
[56] Coe, J. W.; Brooks, P. R. P. US-6410550 B1, 2002.
[57] Busch, F. R.; Hawkins, J. M.; Mustakis, L. G.; Sinay, T. G., Jr.;Watson, T. J. N.; Withbroe, G. J. WO-2006090236 A1, 2006.
PATENT
WO 2002085843
https://google.com/patents/WO2002085843A2?cl=en

PATENT
https://www.google.com/patents/EP2204369A1?cl=en
Varenicline (a compound I of formula I) is the international commonly accepted non-proprietary name for 7,8,9,10-tetrahydro-6,10-methano-6H-pyrazino[2,3-h][3]benzazepine (which is also known as 5,8,14-triazatetracyclo[10.3.1.02,11.04,9]-hexadeca-2(11),3,5,7,9-pentaene), and has an empirical formula of C13H13N3 and a molecular weight of 211.26.

The L-tartrate salt of varenicline is known to be therapeutically useful and is commercially marketed for the treatment of smoking addiction. Varenicline L-tartrate is a partial agonist selective for α4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the trade mark Chantix and is indicated as an aid to smoking cessation treatment.
Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550 . In particular, the preparation of varenicline provided in this reference makes use of 10-aza-tricyclo[6.3.1.02,7]-dodeca-2(7),3,5-triene (a compound of Formula VI), as a key intermediate compound (see Scheme 1 below). Specifically, Example 1 of U.S. Patent No. 6,410,550 describes the synthetic preparation of key intermediate compound of Formula VI as depicted in Scheme 1.

1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III), and / or indane-1,3-dicarbaldehyde (a compound of Formula IV).
Example 1: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)
A 10mL round bottom flask was charged with a compound of formula II (142mg, 1mmol), N-methylmorpholine-N-oxide (120mg, 1.03mmol), tert-butanol (3mL) and water (1mL). FibreCat™ 3003 (OsO4 anchored onto a polymeric support) (11.6mg, 0.0025mmol) was added to this solution and the mixture was heated to reflux. Complete conversion to a compound of formula III was detected by GC, method A, after 48h.
Example 2: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)Step A) Preparation of hexadecyl-trimethylammoniumpermanganate (HTAP):
HTAP was prepared from ion exchange reaction between hexadecyltrimethylammoniumbromide and potassium permanganate.
Potassium permanganate (17.38g, 0.11mol, 1equiv.) was dissolved in 500mL water. A solution of hexadecyltrimethylammoniumbromide (40.10g, 0.11mol, 1equiv) in 500mL water was added drop-wise over 45 min at 20-22°C, and the mixture stirred for 30 minutes at this temperature. The precipitated solid was collected by filtration, washed with water (3 x 100mL) and dried under vacuum at 35°C for 24 hours to give 34.38g of HTAP as a light purple solid.
Step B) Preparation of a compound of formula III:
Compound II (3.52g, 24.8mmol, 1equiv.) was dissolved in anhydrous tetrahydrofuran (80mL) and a solution of HTAP (10g, 24.8mmol, 1.0equiv.) in anhydrous tetrahydrofuran (125mL) was added drop-wise at 23-30°C over 45min. The reaction was monitored by TLC (hexane-ethyl acetate = 1:1). After complete reaction the mixture was cooled to below 10°C, and methyl tert-butyl ether (50mL) and 5% aqueous NaOH solution (50mL) were added and the mixture stirred for 30min. The solid was removed by filtration, and washed with methyl tert-butyl ether (2 x 30mL). The combined layers of the filtrate were separated and the aqueous phase extracted with methyl tert-butyl ether (2 x 30mL). The organic layers were combined and washed with 5% aqueous NaOH solution (50mL), water (2 x 50mL), dried over MgSO4, filtered and concentrated to obtain a dark green solid. This residue was suspended in acetone (15mL) and collected by filtration, washing with additional acetone (3 x 5mL). The product was dried under vacuum at 40°C to give 2.215g (50.7% yield) as a white crystalline solid.
Analytical data: m.p. = 178.8-179.3°C; 1H-NMR: See Figure 1; 13C-NMR: See Figure 2.
Example 3: Preparation of indane-1,3-dicarbaldehyde (a compound of Formula IV)
A 25 mL round bottom flask was charged with a compound of formula I (142mg, 1mmol), Ruthenium (III) chloride hydrate (Aldrich, Reagent Plus™) (7.2mg, 0.035mmol), acetonitrile (8.5mL) and water (1.1mL). The solution was heated to 45°C and sodium periodate (449mg, 2.1mmol) was added portionwise over 25 minutes. After 1h, the reaction was cooled to ambient temperature and filtered. The solids were washed with ethyl acetate (3 x 2mL) and water (3mL). The filtrate was concentrated under vacuum and 5mL of water were added to the obtained residue. The mixture was extracted with ethyl acetate (2 x 5mL) and the combination of the organic layers was washed with water (3 x 5mL), dried with MgSO4 and concentrated under vacuum to obtain a compound of formula IV (118mg) in 68% yield, 70.9% purity (analyzed by GC, method A).
PATENT
WO 199935131, WO 2002092089, US 2013030179

PATENT
https://www.google.com/patents/WO2009065872A2?cl=en
Example 1: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3] benzazepine L-tartrate (i.e. varenicline L-tartrate)
A) Preparation of compound of formula (III)
This example is based on U.S. Patent No. 6,410,550.
A 250 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- triene para-toluene sulfonic acid salt (12.4g, 37.5 mmol) and 44 mL of CH2Cl2. Triethylamine (8.3 g, 82.5 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (8.1q, 41.25 mmol) in 19 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 * 40 mL) and brine (40 mL) . The organic phase was used in the next step without further purification.
On the other hand, a 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (25.9 g, 172.5 mmol), CH2Cl2 (110 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (5.4 g, 86.25 mmol) was added slowly. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (60 mL) an ice (80 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 x 50 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3, dried over Na2SO4 and volatiles evaporated under vacuum to obtain 11.9 g of a solid that was suspended and stirred for 2 hours in AcOEt (12 mL) and hexanes (24 mL) . The solid was filtered and washed with hexanes to obtain the compound of formula (III), 9.1g with a purity of 88.9% by GC (9.8% of meta-dimtrocompound impurity) .
B) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol), damp 5% Pd/C 50% and 180 mL of a 2- propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (40 mL) . To this solution, K2HPO4(458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop- wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the compound of formula (IV), 6.78 g.
C) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International Patent No. WO/2006/090236.
A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with compound of formula (IV) (6.78 g, 22 mmol) and toluene
(47 mL) . To this solution was added a solution of NaOH (2.7 g, 68.2 mmol) in water (34 mL) . The mixture was heated to 400C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (68 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (30 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (90 mL) and evaporated again. The final residue was dissolved in 156 mL of MeOH. 1.3 g of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain an intense yellow solution. The process with activated carbon was repeated without any improvement in the colour. This solution was added drop-wise over a solution of L- tartaric acid (3.63 g, 24.2 mmol) in MeOH (47 mL) . The slurry was stirred for 72 hours at room temperature, filtered, washed with MeOH and dried in an oven at 50 0C for 8 hours, to obtain 5.05 g of varenicline L-tartrate as a yellow solid with a 95.5% purity by HPLC (4.4% of unknown impurity A). Colour L: 92.75, a*: -7.19, b*:43.08.
Comparative Example 2: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazmo [2, 3-h] [3 ] benzazepine L-tartrate (i.e. varenicline L-tartrate) A) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) prepared according to Comparative Example 1.A) (4.1 g) , 123 mg of damp 5% Pd/C 50% and 81 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 24 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (16 mL) . To this solution, K2HPO4 (207 mg, 1.19 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 1.84 g of 40% aqueous glyoxal diluted with water (6.6 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 30 mL and water (56 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water and dried in a oven at 50 0C to obtain 3.15 g of compound of formula (IV) .
B) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International application No. WO/2006/090236. A 100 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with
7, 8, 9, 10-tetrahydro-8- (tπfluoroacetyl) -6, 10-methano-6H- pyrazino [2 , 3-h] [3] benzazepine, i.e. compound of formula
(IV) (3.14 g, 10.2 mmol) and toluene (22 mL) . To this solution was added a solution of NaOH (1.3 g, 31.6 mmol) in water (16 mL) . The mixture was heated to 40 0C and stirred for 2.5 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (30 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (15 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (45 mL) and evaporated again. The final residue was dissolved m 70 mL of MeOH. 314 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 mm and filtered through Celite to obtain a yellow solution. This solution was added drop-wise over a solution of L- tartaπc acid (1.68 g, 11.22 mmol) m MeOH (22 mL) . The slurry was stirred for 1 hour at room temperature, filtered, washed with MeOH (2 x 5 mL) and dried under vacuum, to obtain vareniclme L-tartrate (2.48 g) as a yellow solid with a 95.6% purity by HPLC (4.4% of unknown impurity A). Colour L: 99.50, a*: -4.98, b*:43.02
Comparative Example 3: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3 ] benzazepine L-tartrate (i.e. vareniclme L-tartrate)
This example is based on International application No. WO/2002/092089.
2 g of vareniclme L-tartrate as obtained from Comparative Example 1 were dissolved in 3 mL of water.
To this solution, 100 mL of CH3CN were added, and the resulting slurry was stirred for 10 mm and filtered.
After drying the product was analysed to be a 98.2% purity by HPLC (1.7% of unknown impurity A) . Colour L: 91.44, a*: -3.24, b* : 33.47
Example 1: Preparation of 7, 8, 9, lO-tetrahydro-6, 10- methano-6H-pyrazmo [2, 3-h] [3] benzazepine L-tartrate
(i.e. vareniclme L-tartrate)
A) Preparation of compound of formula (III) This example is based on U.S. Patent No. 6,410,550, except for the purification step, which is the object of the present invention (i.e. crystallization in toluene) .
A 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- tπene para-toluene sulfonic acid salt (32.5g, 98.2 mmol) and 115 mL of CH2Cl2. Triethylamine (21.8 g, 216 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (22.7 g, 108 mmol) in 50 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 x 100 mL) and brine (100 mL) . The organic phase was used in the next step without further purification.
A l L round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (67.8 g, 452 mmol), CH2Cl2 (280 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (14.2 g, 226 mmol) was slowly added. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (150 mL) an ice (200 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (100 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3 (2×100 mL) , water (100 mL) , dried over Na2SO4 and volatiles evaporated under vacuum to obtain 30.5 g of a solid with a 83.6% purity by GC (12.5% of meta- dinitrocompound impurity) . 20 g of this solid were crystallized in toluene (100 mL) to obtain the compound of formula (III), 15 g of a pale brown solid with a 98.5 % purity by GC (meta-dinitrocompound impurity not detected) .
B) Preparation of compound of formula (IV) This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol, crystals from toluene), damp 5% Pd/C 50% and 180 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered over Celite and washed with 2- propanol (40 mL) . To this solution, K2HPO4 (458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the product, 7.16 g of compound of formula (IV) with a 99.9% purity by HPLC. C) Preparation of varenicline L-tartrate (compound of formula ( I) )
Thrs example rs based on International Patent No. WO/2006/090236. A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with a solution of NaOH (2.89 g, 72.23 mmol) in water (36 mL) , compound of formula (IV) (7.15 g, 23.3 mmol) and toluene (50 mL) . The mixture was heated to 40 0C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (71 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (36 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (110 mL) and evaporated again. The final residue was dissolved in 164 mL of MeOH. 750 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain a yellow solution. This solution was added drop- wise over a solution of L-tartaric acid (3.84 g, 25.6 mmol) in MeOH (50 mL) . The slurry was stirred for 14 hours at room temperature, filtered, washed with MeOH and dried under vacuum, to obtain varenicline L-tartrate
(7.04 g) as an off-white solid with a >99.9% purity by HPLC (unknown impurity A not detected) . Colour L: 94.39, a*: 2.27, b*:9.02.

Post-marketing surveillance
No evidence for increased risks of cardiovascular events, depression, or self-harm with varenicline versus nicotine replacement therapy has been found in one post-marketing surveillance study.[23]
Mechanism of action
Varenicline displays full agonism on α7 nicotinic acetylcholine receptors.[24][25] And it is a partial agonist on the α4β2, α3β4, and α6β2 subtypes.[26] In addition, it is a weak agonist on the α3β2 containing receptors.
Varenicline’s partial agonism on the α4β2 receptors rather than nicotine’s full agonism produces less effect of dopamine release than nicotine’s. This α4β2 competitive binding, reduces the ability of nicotine to bind and stimulate the mesolimbic dopamine system – similar to the method of action of buprenorphine in the treatment of opioid addiction.[3]
Pharmacokinetics
Most of the active compound is excreted by the kidneys (92–93%). A small proportion is glucuronidated, oxidised, N-formylated or conjugated to a hexose.[27] The elimination half-life is about 24 hours.
History
Use of Cytisus plant as a smoking substitute during World War II[28] led to use as a cessation aid in eastern Europe and extraction of cytisine.[29] Cytisine analogs led to varenicline at Pfizer.[30][31][32]
Varenicline received a “priority review” by the US FDA in February 2006, shortening the usual 10-month review period to 6 months because of its demonstrated effectiveness inclinical trials and perceived lack of safety issues.[33] The agency’s approval of the drug came on May 11, 2006.[4] On August 1, 2006, varenicline was made available for sale in the United States and on September 29, 2006, was approved for sale in the European Union.[34]
SEE

| US6410550 | Nov 13, 1998 | Jun 25, 2002 | Pfizer Inc | Aryl fused azapolycyclic compounds |
| WO2009155403A2 * | Jun 18, 2009 | Dec 23, 2009 | Teva Pharmaceutical Industries Ltd. | Processes for the preparation of varenicline and intermediates thereof |
| Reference | ||
|---|---|---|
| 1 | * | BHUSHAN, VIDYA; RATHORE, RAJENDRA; CHANDRASEKARAN, S.: “A Simple and Mild Method for the cis-Hydroxylation of Alkenes with Cetyltrimethylammonium Permanganate” SYNTHESIS, no. 5, 1984, pages 431-433, XP002581198 |
| 2 | * | BROOKS P R ET AL: “Synthesis of 2,3,4,5-tetrahydro-1,5-methano-1H-3-benzaz epine via oxidative cleavage and reductive amination strategies” SYNTHESIS 20040803 DE, no. 11, 3 August 2004 (2004-08-03), pages 1755-1758, XP002581197 ISSN: 0039-7881 |
| 3 | * | SORBERA L A ET AL: “Varenicline tartrate: Aid to smoking cessation nicotinic [alpha]4[beta]2 partial agonist” DRUGS OF THE FUTURE 200602 ES LNKD- DOI:10.1358/DOF.2006.031.02.964028, vol. 31, no. 2, February 2006 (2006-02), pages 117-122, XP002581199 ISSN: 0377-8282 DOI: 10.1358/dof.2006.031.02.964028 |
| WO2001062736A1 * | Feb 8, 2001 | Aug 30, 2001 | Pfizer Products Inc. | Aryl fused azapolycyclic compounds |
| WO2002085843A2 * | Mar 4, 2002 | Oct 31, 2002 | Pfizer Products Inc. | Process for the preparation of 1,3-substituted indenes and aryl-fused azapolycyclic compounds |
| WO2006090236A1 * | Feb 21, 2006 | Aug 31, 2006 | Pfizer Products Inc. | Preparation of high purity substituted quinoxaline |
| WO2008060487A2 * | Nov 9, 2007 | May 22, 2008 | Pfizer Products Inc. | Polymorphs of nicotinic intermediates |
| Reference | ||
|---|---|---|
| 1 | * | COE J W ET AL: “Varenicline: an alpha4beta2 Nicotinic Receptor Partial Agonist for Smoking Cessation” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON., US, vol. 48, no. 10, 1 January 2005 (2005-01-01), pages 3474-3477, XP002474642 ISSN: 0022-2623 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010005643A1 * | May 28, 2009 | Jan 14, 2010 | Teva Pharmaceutical Industries Ltd. | Processes for purifying varenicline l-tartrate salt and preparing crystalline forms of varenicline l-tartrate salt |
| WO2011110954A1 * | Mar 8, 2011 | Sep 15, 2011 | Actavis Group Ptc Ehf | Highly pure varenicline or a pharmaceutically acceptable salt thereof substantially free of methylvarenicline impurity |
| WO2011154586A3 * | Jun 13, 2011 | Mar 22, 2012 | Medichem, S. A. | Improved methods for the preparation of quinoxaline derivatives |
| EP2581375A2 * | Jun 13, 2011 | Apr 17, 2013 | Medichem, S.A. | Improved methods for the preparation of quinoxaline derivatives |
| US8039620 | May 21, 2009 | Oct 18, 2011 | Teva Pharmaceutical Industries Ltd. | Varenicline tosylate, an intermediate in the preparation process of varenicline L-tartrate |
| US8178537 | Jun 22, 2010 | May 15, 2012 | Teva Pharmaceutical Industries Ltd. | Solid state forms of varenicline salts and processes for preparation thereof |
References
- Jump up^ Mills EJ, Wu P, Spurden D, Ebbert JO, Wilson K (2009). “Efficacy of pharmacotherapies for short-term smoking abstinance: a systematic review and meta-analysis” (PDF). Harm Reduct J 6: 25. doi:10.1186/1477-7517-6-25. PMC 2760513. PMID 19761618.
- ^ Jump up to:a b Cahill K, Stevens S, Perera R, Lancaster T (May 2013). “Pharmacological interventions for smoking cessation: an overview and network meta-analysis”. Cochrane Database Syst Rev (Systematic Review & Meta-Analysis) 5: CD009329.doi:10.1002/14651858.CD009329.pub2. PMID 23728690.
- ^ Jump up to:a b c d Elrashidi MY, Ebbert JO (June 2014). “Emerging drugs for the treatment of tobacco dependence: 2014 update”. Expert Opin Emerg Drug (Review) 19 (2): 243–60.doi:10.1517/14728214.2014.899580. PMID 24654737.
- ^ Jump up to:a b U.S. Food and Drug Administration.FDA Approves Novel Medication for Smoking Cessation. Press release, 11 May 2006.
- Jump up^ Cressman, AM; Pupco, A; Kim, E; Koren, G; Bozzo, P (May 2012). “Smoking cessation therapy during pregnancy.”. Canadian Family Physician 58 (5): 525–7. PMC 3352787.PMID 22586193.
- Jump up^ “Varenicline Pregnancy Cohort Study”. clinicaltrials.gov.
- Jump up^ “LactMed”. nih.gov.
- Jump up^ Leung, LK; Patafio, FM; Rosser, WW (September 28, 2011). “Gastrointestinal adverse effects of varenicline at maintenance dose: a meta-analysis”. BMC clinical pharmacology11 (1): 15. doi:10.1186/1472-6904-11-15. PMC 3192741. PMID 21955317.
- American Cancer Society. “Cancer Drug Guide: Varenicline”. Retrieved 2008-01-19.
- Jump up^ “DailyMed – CHANTIX- varenicline tartrate”. nih.gov.
- FDA. “Public Health Advisory: FDA Requires New Boxed Warnings for the Smoking Cessation Drugs Chantix and Zyban”. Retrieved 2009-07-01.
- ^ Jump up to:a b “www.accessdata.fda.gov” (PDF).
- Hughes, JR (8 January 2015). “Varenicline as a Cause of Suicidal Outcomes.”. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco.doi:10.1093/ntr/ntu275. PMID 25572451.
- “FDA Drug Safety Communication: Chantix (varenicline) may increase the risk of certain cardiovascular adverse events in patients with cardiovascular disease”. 2011-06-16.
- Jump up^ Singh, S; Loke, YK, Spangler, JG, Furberg, CD (Sep 6, 2011). “Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis” (PDF). CMAJ : Canadian Medical Association 183 (12): 1359–66.doi:10.1503/cmaj.110218. PMC 3168618. PMID 21727225.
- Takagi, H; Umemoto, T (Sep 6, 2011). “Varenicline: quantifying the risk”. CMAJ : Canadian Medical Association 183 (12): 1404. doi:10.1503/cmaj.111-2063.PMC 3168634. PMID 21896705.
- Jump up^ Samuels, L (Sep 6, 2011). “Varenicline: cardiovascular safety”. CMAJ : Canadian Medical Association 183 (12): 1407–08. doi:10.1503/cmaj.111-2073. PMC 3168639.PMID 21896709.
- “European Medicine Agency confirms positive benefit-risk balance for Champix.”. 2011-07-21.
- ^ Jump up to:a b Prochaska JJ, Hilton JF (2012). “Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis”. BMJ (Systematic Review & Meta-Analysis) 344: e2856.doi:10.1136/bmj.e2856. PMC 3344735. PMID 22563098.
- Mills EJ, Thorlund K, Eapen S, Wu P, Prochaska JJ (January 2014). “Cardiovascular events associated with smoking cessation pharmacotherapies: a network meta-analysis”.Circulation (Network Meta-Analysis) 129 (1): 28–41.doi:10.1161/CIRCULATIONAHA.113.003961. PMID 24323793.
- cessation in cardiovascular patients”. Evidence-Based Medicine (Review & Commentary) 19 (5): 193. doi:10.1136/eb-2014-110030.PMID 24917603.
- Rowland K (April 2014). “ACP Journal Club. Review: Nicotine replacement therapy increases CVD events; bupropion and varenicline do not”. Annals of Internal Medicine(Review & Commentary) 160 (8): JC2. doi:10.7326/0003-4819-160-8-201404150-02002.PMID 24733219.
- Jump up^ Kotz D, Viechtbauer W, Simpson C, van Schayck OC, West R, Sheikh A (2015).“Cardiovascular and neuropsychiatric risks of varenicline: a retrospective cohort study”.Lancet Respir Med (retrospective cohort) 3: 761–768. doi:10.1016/S2213-2600(15)00320-3. PMC 4593936. PMID 26355008.
- Jump up^ Mihalak KB, Carroll FI, Luetje CW; Carroll; Luetje (2006). “Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors”. Mol. Pharmacol.70 (3): 801–805. doi:10.1124/mol.106.025130. PMID 16766716.
- Jump up^ Mineur YS, Picciotto MR; Picciotto (December 2010). “Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis”. Trends Pharmacol. Sci. 31 (12): 580–6. doi:10.1016/j.tips.2010.09.004. PMC 2991594. PMID 20965579.
- Tanuja Bordia. “Varenicline Is a Potent Partial Agonist at α6β2* Nicotinic Acetylcholine Receptors in Rat and Monkey Striatum”. aspetjournals.org.
- Obach, RS; Reed-Hagen, AE; Krueger, SS; Obach, BJ; O’Connell, TN; Zandi, KS; Miller, S; Coe, JW (2006). “Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro”. Drug metabolism and disposition: the biological fate of chemicals 34 (1): 121–130.doi:10.1124/dmd.105.006767. PMID 16221753.
- “[Cytisine as an aid for smoking cessation].”. Med Monatsschr Pharm 15 (1): 20–1. Jan 1992. PMID 1542278.
- Prochaska, BMJ 347:f5198 2013 http://www.bmj.com/content/347/bmj.f5198
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD 3rd, O’Neill BT (2005). “Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation”. J. Med. Chem. 48(10): 3474–3477. doi:10.1021/jm050069n. PMID 15887955.
- Schwartz JL (1979). “Review and evaluation of methods of smoking cessation, 1969–77. Summary of a monograph”. Public Health Rep 94 (6): 558–63. PMC 1431736.PMID 515342.
- Etter JF (2006). “Cytisine for smoking cessation: a literature review and a meta-analysis”. Arch. Intern. Med. 166 (15): 1553–1559. doi:10.1001/archinte.166.15.1553.PMID 16908787.
- Kuehn BM (2006). “FDA speeds smoking cessation drug review”. JAMA 295 (6): 614–614.doi:10.1001/jama.295.6.614. PMID 16467225.
- European Medicines Agency (2011-01-28). “EPAR summary for the public. Champix varenicline”. London. Retrieved 2011-02-14.
External links
Manufacturer’s website USA

|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7,8,9,10-Tetrahydro-6,10-methano-6H-pyrazino[2,3-h] [3]benzazepine
|
|
| Clinical data | |
| Trade names | Chantix |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a606024 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | <20% |
| Metabolism | Limited (<10%) |
| Biological half-life | 24 hours |
| Excretion | Renal (81–92%) |
| Identifiers | |
| CAS Number | 249296-44-4  375815-87-5 375815-87-5 |
| ATC code | N07BA03 (WHO) |
| PubChem | CID 5310966 |
| IUPHAR/BPS | 5459 |
| DrugBank | DB01273 |
| ChemSpider | 4470510 |
| UNII | W6HS99O8ZO |
| KEGG | D08669  |
| ChEBI | CHEBI:84500 |
| ChEMBL | CHEMBL1076903 |
| Chemical data | |
| Formula | C13H13N3 |
| Molar mass | 211.267 g/mol |
////////////Varenicline, Chantix™, FDA 2006, 249296-44-4, 375815-87-5, Champix , Pfizer, バレニクリン酒石酸塩
n1c2cc3c(cc2ncc1)[C@@H]4CNC[C@H]3C4
Filed under: Uncategorized Tagged: 249296-44-4, 375815-87-5, バレニクリン酒石酸塩, Champix, Chantix™, FDA 2006, PFIZER, Varenicline







