
Eliglustat tartrate (Cerdelga) エリグルスタット酒石酸塩
依利格鲁司特
エリグルスタット,サーデルガ
FOR TREATMENT OF GAUCHERS DISEASE

ELIGLUSTAT; Cerdelga; Genz 99067; Genz-99067; UNII-DR40J4WA67; GENZ-112638;
CAS 491833-29-5 FREE FORM
| Molecular Formula: | C23H36N2O4 |
|---|---|
| Molecular Weight: | 404.54294 g/mol |
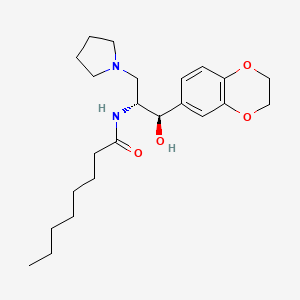
N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide;(2R,3R)-2,3-dihydroxybutanedioic acid
Mechanism of Action: glucosylceramide synthase inhibitor
Indication: Type I Gaucher Disease
Date of Approval: August 19, 2014 (US)
US patent number:US6916802 , US7196205 , US7615573
Patent Expiration Date: Apr 29, 2022 (US6916802, US7196205, US7615573)
Exclusivity Expiration Date:Aug 19, 2019(NCE), Aug 19, 2021 (ODE)
Originator:University of Michigan
Developer: Genzyme, a unit of Sanofi
Eliglustat, marketed by Genzyme as CERDELGA, is a glucosylceramide synthase inhibitor indicated for the long-term treatment of type 1 Gaucher disease. Patients selected for treatment with Eliglustat undergo an FDA approved genotype test to establish if they are CYP2D6 EM (extensive metabolizers), IM (intermediate metabolizers), or PM (poor metabolizers), as the results of this test dictate the dosage of Eliglustat recommended. Eliglustat was approved for use by the FDA in August 2014.
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s disease developed by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4] According to an article in Journal of the American Medical Association the oral substrate reduction therapy resulted in “significant improvements in spleen volume, hemoglobin level, liver volume, and platelet count” in untreated adults with Gaucher disease Type 1.[5]
Cerdelga, capsule, 84 mg/1, oralGenzyme Corporation, 2014-09-03, 

Eliglustat tartrate
- Molecular FormulaC50H78N4O14
- Average mass959.173 Da
- UNII-N0493335P3
-
Butanedioic acid, 2,3-dihydroxy-, (2R,3R)-, compd. with N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-pyrrolidinylmethyl)ethyl]octanamide (1:2)
-
eliglustat hemitartrate
-
eliglustat L-tartrate
CAS 928659-70-5
![]()


CERDELGA (eliglustat) capsules contain eliglustat tartrate, which is a small molecule inhibitor of glucosylceramide synthase that resembles the ceramide substrate for the enzyme, with the chemical name N-((1R,2R)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1- hydroxy-3-(pyrrolidin-1-yl)propan-2-yl)octanamide (2R,3R)-2,3-dihydroxysuccinate. Its molecular weight is 479.59, and the empirical formula is C23H36N2O4+½(C4H6O6) with the following chemical structure:
|
Each capsule of CERDELGA for oral use contains 84 mg of eliglustat, equivalent to 100 mg of eliglustat tartrate (hemitartrate salt). The inactive ingredients are microcrystalline cellulose, lactose monohydrate, hypromellose and glyceryl behenate, gelatin, candurin silver fine, yellow iron oxide, and FD&C blue 2.
Cost
In 2014, the annual cost of Cerdelga hard gelatin capsules taken orally twice a day was $310,250. Genzyme’s flagship Imiglucerase(brand name Cerezyme) cost about $300,000 for the infusions if taken twice a month.[6] Manufacturing costs for Cerdelga are slightly lower than for Cerezyme. Genzyme’s maintains higher prices for orphan drugs—most often paid for by insurers— in order to remain financially sustainable.[6]
Chemically Eliglustat is named N-[(1 R,2R)-2-(2,3-dihydro-1 ,4-benzodioxin-6-yl)-2-hydroxy-1 -(1 -pyrrolidinylmethyl)ethyl]-Octanamide(2R!3R)-2,3-dihydroxybutanedioate and the hemitartarate salt of eliglustat has the structural formula as shown in Formula I.

Formula I
Eliglustat hemitartrate (Genz-1 12638), currently under development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase, and is currently under development by Genzyme.
U.S. patent No. 7,196,205 discloses a process for the preparation of Eliglustat or a pharmaceutically acceptable salt thereof.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 discloses process for preparation of Eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of Eliglustat, (ii) a hemitartrate salt of Eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It has been disclosed earlier that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailablity patterns compared to crystalline forms [Konne T., Chem pharm Bull., 38, 2003(1990)]. For some therapeutic indications one bioavailabihty pattern may be favoured over another. An amorphous form of Cefuroxime axetil is a good example for exhibiting higher bioavailability than the crystalline form.
CLIP
Eliglustat tartrate, developed and marketed by Genzyme Corporation (a subsidiary of Sanofi), was approved by the US FDA in August 2014 for the treatment of nonneuropathic (type 1) Gaucher disease (GD1) in both treatment-naïve and treatment-experienced adult patients.98
It is the first oral treatment to be approved for first-line use in patients with Gaucher disease type 1, which is a rare lysosomal storage disease characterized by accumulation of lipid glucosylceramide (GL-1) due to insufficient production of the enzyme glucosylceramidase.99,100
Clinical complications include hepatosplenomegaly, anemia, thrombocytopenia, and bone involvement.101 Eliglustat is a specific inhibitor of glucosylceramide synthase with an IC50 of 10 ng/mL and acts as substrate reduction therapy for GD1;102 it has demonstrated non-inferiority to enzyme replacement therapy, which is the current standard of care, in Phase III trials.99
While the process-scale route has not yet been disclosed,103 the largest scale route to eliglustat tartrate reported to date is described in Scheme 15.104
Condensation of commercially available S-(+)-2-phenyl glycinol (87) with phenyl bromoacetate (88) in acetonitrile in the presence of N,N-diisopropylethylamine (DIPEA) provided morpholin-2-one 89 upon treatment with HCl.Neutralization with NaHCO3 followed by coupling with aldehyde 90 in refluxing EtOAc/toluene yielded oxazine adduct 91, which was isolated as a precipitate from methyl-tert-butyl ether (MTBE).
The stereochemistry of the three new stereocenters in 91 can be rationalized through the cycloaddition of an ylide intermediate in the sterically-preferred S-configuration (generated by the reaction of the morpholinone 89 with aldehyde 90) with a second equivalent of the aldehyde. With the morpholinone in a chair conformation in which the phenyl group is equatorial, endo axial approach of the dipolarophile to the less-hindered face of the ylide and subsequent ring flip of the morpholinone ring to a boat conformation positions all exocyclic aryl substituents in a pseudoequatorial configuration. 105
Opening of oxazine 91 with pyrrolidine in refluxing THF followed by addition of HCl in refluxing MeOH gave amide 92, which was reduced to amine 93 using LiAlH4 in refluxing THF.
Subsequent hydrogenation with Pd(OH)2 in EtOH cleaved the phenylethanol group to give the free amine, which was converted to dioxalate salt 94 by treatment with oxalic acid in methyl isobutylketone (MIBK). Subjection of aminoethanol 94 to aqueous sodium hydroxide followed by coupling with palmitic acid Nhydroxysuccinimide (NHS)-ester (95) gave eliglustat as the corresponding freebase (96) in 9.5% overall yield from 87.
Salt formation with L-tartaric acid (0.5 equiv) then provided eliglustat tartrate (XII).106


98. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm410585.htm.
99. Poole, R. M. Drugs 2014, 74, 1829.
100. Kaplan, P. Res. Rep. Endocr. Disord. 2014, 4, 1.
101. Pastores, G. M.; Hughes, D. Clin. Invest. 2014, 4, 45.
102. Shayman, J. A. Drugs Future 2010, 35, 613.
103. Javed, I.; Dahanukar, V. H.; Oruganti, S.; Kandagatla, B. WO Patent2,015,059,679, 2015.
104. Hirth, B.; Siegel, C. WO Patent 2,003,008,399, 2003.
105. Anslow, A. S.; Harwood, L. M.; Phillips, H.; Watkin, D.; Wong, L. F. Tetrahedron:Asymmetry 1991, 2, 1343.
106. Liu, H.; Willis, C.; Bhardwaj, R.; Copeland, D.; Harianawala, A.; Skell, J.;Marshall, J.; Kochling, J.; Palace, G.; Peterschmitt, J.; Siegel, C.; Cheng, S. WO Patent 2,011,066,352, 2011.
CLIP
TAKEN FROM
http://www.xinbiaopin.com/a/zuixindongtai/huaxuepinshuju/2015/0310/2383.html


Nmr predict
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide H-NMR spectrum](http://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-1h.png)
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](http://i1.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis


PATENT
http://www.google.com/patents/WO2013059119A1?cl=en

http://www.google.com/patents/US7196205
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide

This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol

Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol

To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4 (Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).
PATENT
EXAMPLES
Example 1 : Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 14 mL of dichloromethane at 26°C and stirred for 15 min. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 45°C. After distillation the solid was dried under vacuum at 45°C.
PATENT

PAPER
Journal of Medicinal Chemistry (2012), 55(9), 4322-4335
OLD CLIPS
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease

Eliglustat tartrate (USAN)
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.

Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)-3-hydroxy-2-(2”-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2(40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
References
- Eligustat (PDF), AMA By subscription only
- FDA approves new drug to treat a form of Gaucher disease, U.S. Food and Drug Administration, 19 August 2015, retrieved 18 July 2015
- Lee, L.; Abe, A.; Shayman, J. A. (21 May 1999). “Improved Inhibitors of Glucosylceramide Synthase”. Journal of Biological Chemistry 274(21): 14662–14669. doi:10.1074/jbc.274.21.14662.
- Shayman, JA (1 August 2010). “Eliglustat Tartrate: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease.”. Drugs of the future 35 (8): 613–620. PMID 22563139.
- Pramod K. Mistry, Elena Lukina, Hadhami Ben Turkia, Dominick Amato, Hagit Baris, Majed Dasouki, Marwan Ghosn, Atul Mehta, Seymour Packman, Gregory Pastores, Milan Petakov, Sarit Assouline, Manisha Balwani, Sumita Danda, Evgueniy Hadjiev, Andres Ortega, Suma Shankar, Maria Helena Solano, Leorah Ross, Jennifer Angell, M. Judith Peterschmitt (17 February 2015), “Effect of Oral Eliglustat on Splenomegaly in Patients With Gaucher Disease Type 1: The ENGAGE Randomized Clinical Trial”, Journal of the American Medical Association 313 (7): 695–706, doi:10.1001/jama.2015.459
- Robert Weisman (2 September 2014), New Genzyme pill will cost patients $310,250 a year, The Boston Globe, retrieved 18 July 2015
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 | |
|---|---|
| Patent | 6916802 |
| Expiration | Apr 29, 2022 |
| Applicant | GENZYME CORP |
| Drug Application | N205494 (Prescription Drug: CERDELGA. Ingredients: ELIGLUSTAT TARTRATE) |
| FDA Orange Book Patents: 2 of 3 | |
|---|---|
| Patent | 7196205 |
| Expiration | Apr 29, 2022 |
| Applicant | GENZYME CORP |
| Drug Application | N205494 (Prescription Drug: CERDELGA. Ingredients: ELIGLUSTAT TARTRATE) |
| Patent ID | Date | Patent Title |
|---|---|---|
| US8003617 | 2011-08-23 | Methods of Treating Diabetes Mellitus |
| US2010298317 | 2010-11-25 | METHOD OF TREATING POLYCYSTIC KIDNEY DISEASES WITH CERAMIDE DERIVATIVES |
| US7763738 | 2010-07-27 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYLTRANSFERASE INHIBITORS |
| US7615573 | 2009-11-10 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US2009105125 | 2009-04-23 | Methods of Treating Fatty Liver Disease |
| US7265228 | 2007-09-04 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US7196205 | 2007-03-27 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US6855830 | 2005-02-15 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016068519 | 2016-03-10 | INHIBITORS OF THE ENZYME UDP-GLUCOSE: N-ACYL-SPHINGOSINE GLUCOSYLTRANSFERASE |
| US2015148534 | 2015-05-28 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYL TRANSFERASE INHIBITORS |
| US2015051261 | 2015-02-19 | Methods of Treating Fatty Liver Disease |
| US8779163 | 2014-07-15 | Synthesis of UDP-Glucose: N-acylsphingosine glucosyl transferase inhibitors |
| US2013137743 | 2013-05-30 | AMORPHOUS AND A CRYSTALLINE FORM OF GENZ 112638 HEMITARTRATE AS INHIBITOR OF GLUCOSYLCERAMIDE SYNTHASE |
| US2013095089 | 2013-04-18 | GLUCOSYLCERAMIDE SYNTHASE INHIBITORS AND THERAPEUTIC METHODS USING THE SAME |
| US2012322786 | 2012-12-20 | 2-ACYLAMINOPROPOANOL-TYPE GLUCOSYLCERAMIDE SYNTHASE INHIBITORS |
| US8138353 | 2012-03-20 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYLTRANSFERASE INHIBITORS |
| US2012022126 | 2012-01-26 | Method Of Treating Diabetes Mellitus |
| US8003617 | 2011-08-23 | Methods of Treating Diabetes Mellitus |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide
|
|
| Clinical data | |
| Trade names | Cerdelga |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 491833-29-5 |
| ATC code | A16AX10 (WHO) |
| PubChem | CID 23652731 |
| ChemSpider | 28475348 |
| ChEBI | CHEBI:82752  |
| Chemical data | |
| Formula | C23H36N2O4 |
| Molar mass | 404.543 g/mol |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US6916802 | No | 2002-04-29 | 2022-04-29 | |
| US7196205 | No | 2002-04-29 | 2022-04-29 | |
| US7615573 | No | 2002-04-29 | 2022-04-29 | |
///////////491833-29-5, 928659-70-5, eliglustat hemitartrate, eliglustat L-tartrate, ELIGLUSTAT, Cerdelga, Genz 99067, Genz-99067, UNII-DR40J4WA67, GENZ-112638, エリグルスタット酒石酸塩 , FDA 2014, GAUCHERS DISEASE, 依利格鲁司特, エリグルスタット,サーデルガ
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@@H](C2=CC3=C(C=C2)OCCO3)O
Filed under: FDA 2014 Tagged: 491833-29-5, 928659-70-5, エリグルスタット, エリグルスタット酒石酸塩, サーデルガ, Cerdelga, eliglustat, eliglustat hemitartrate, eliglustat L-tartrate, FDA 2014, GAUCHERS DISEASE, Genz 99067, GENZ-112638, UNII-DR40J4WA67, 依利格鲁司特







