
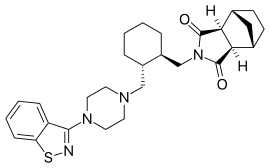
Lurasidone – it having been developed and launched by Sumitomo Dainippon Pharma. Lurasidone was launched for schizophrenia in the US by Sumitomo’s US subsidiary Sunovion Pharmaceuticals.
WO 2016110798, Piramal Enterprises Ltd, New Patent, Lurasidone
An improved process for the preparation of lurasidone and its intermediate
PIRAMAL ENTERPRISES LIMITED [IN/IN]; Piramal Tower Ganpatrao Kadam Marg, Lower Parel Mumbai 400013 (IN)

GHARPURE, Milind; (IN).
TIWARI, Shashi Kant; (IN).
WAGH, Ganesh; (IN).
REVANAPPA, Galge; (IN).
WARPE, Manikrao; (IN).
ZALTE, Yogesh; (IN).

From left: Anand Piramal, executive director, Piramal Group; Swati Piramal, vice-chairperson, Piramal Group; Ajay Piramal, chairman, Piramal Group; Nandini Piramal, executive director, Piramal Enterprises; and Peter DeYoung, president, Piramal Enterprises
Improved process for preparing pure (3aR,7aR)-4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2,1′-piperazin]-1′-ium methanesulfonate, useful as a key intermediate in the synthesis of lurasidone. Also claims a process for purifying lurasidone hydrochloride, useful for treating schizophrenia and bipolar disorders. In July 2016, Newport Premium™ reported that Piramal Enterprises was capable of producing commercial quantities of lurasidone hydrochloride and holds an active US DMF for the drug since March 2015.
Lurasidone (the Compound-I), is an atypical antipsychotic used in the treatment of schizophrenia and bipolar disorders.The drug is marketed as hydrochloride salt (the compound-I.HCl) by Sunovion Pharms Inc.under the tradename”LATUDA”, in the form of oral tablets. Latuda is indicated for the treatment of patients with schizophrenia. Lurasidone hydrochloride has the chemical name ((3aR,4S,7R,7aS)-2-[((lR,2R)-2-{ [4-(l,2-benzisothiazol-3-yl)-piperazin-l-yl]methyl}cyclohexyl)-methyl]hexahydro-lH-4,7-methanisoindol-l,3-dione hydrochloride, and is structurally represented as follows;

Compound-I.HCl
Lurasidone being an important antipsychotic agent; a number of processes for its preparation as well as for its intermediates are known in the art.
US Patent No. 5,532,372 describe a process for the synthesis of Lurasidone, which is illustrated below in Scheme-I. In the process, the compound, cyclohexane- l,2-diylbis(methylene) dimethanesulfonate(referred to as the compound-Ill) is reacted with 3-(l-piperazinyl-l,2-benzisothiazole(referred to as the compound-IV) in acetonitrile, and in the presence of sodium carbonate to provide corresponding quaternary ammonium salt as 4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2, r-piperazin]-l’-ium methanesulfonate (the compound-II). The compound-II is further treated with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in xylene, in the presence of potassium carbonate and dibenzo-18-crown-6-ether to provide lurasidone.

Scheme-I
US Published Patent Application 2011/0263848 describes a process for the preparation of the quaternary ammonium salt (the compound-II) which comprises reacting 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)- cyclohexane in a solvent such as toluene in the presence of a phosphate salt.
Indian Published Patent Application 2306/MUM/2014 (” the IN’2306 Application”) describes a process for the synthesis of lurasidone and the intermediates thereof, comprising reacting (R,R) trans l,2-bis(methane sulphonyl methyl)cyclohexane with 3-(Piperazine-l-yl)benzo[d]isothiazole in presence of a mixture of two or more polar aprotic solvents selected from acetonitrile, N,N-dimethyl formamide (DMF) and/or Ν,Ν-dimethyl acetamide (DMAc), and a base at reflux temperature to obtain the quaternary ammonium salt (the compound II), which is then converted to lurasidone. The IN’2306 application demonstrated preparation of the compound II using the solvent combination such as acetonitrile-DMF and acetonitrile-DMAc.
US Published Patent Application 2011/0263847 describes a process for the preparation of the quaternary ammonium salt (the compound-II) comprising reacting 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent such as toluene, wherein the piperazine compound is used in an excess amount i.e. 1.8 to 15 moles with respect to ( 1R,2R)- 1 ,2-bis(methanesulfonyloxymethyl)cyclohexane.
Chinese Published Patent Application 102731512 describes a process for the preparation of the quaternary ammonium salt (the compound-II) comprises reaction of 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent such as toluene in the presence of a phase transfer catalyst.
In addition to the afore discussed patent documents, there are a number of patent documents that describe a process for the preparation of the quaternary ammonium salt (the compound-II), the key intermediate for the synthesis of lurasidone. For instance, Published PCT application WO2012/131606 A 1, Indian Published patent application 217/MUM/2013, Chinese published patent applications 102863437, 103864774 and 102827157 describe a process for the preparation of the quaternary ammonium salt (compound-II) comprises reaction of 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent or a solvent mixture such as acetonitrile, acetonitrile : water solvent mixture, toluene or DMF, in the presence of a base.
It is evident from the discussion of the processes for the preparation of the quaternary ammonium salt (the compound-II), described in the afore cited patent documents that the reported processes primarily involve use of acetonitrile either as the single solvent or in a mixture of solvents. Acetonitrile is a relatively toxic, and not an environment friendly solvent. Due to its toxic nature, it can cause adverse health effects also. Acetonitrile is covered under Class 2 solvents i.e. solvents to be limited, and residual solvent limit of acetonitrile is 410 ppm in a drug substance as per the ICH (International Conference on Harmonisation) guidelines for residual solvents. Moreover, acetonitrile is a costlier solvent, which renders the process costlier and hence, is not an industrially feasible solvent.
It is also evident from the discussion of the processes described in afore cited patent documents that some of the reported processes involve use of high boiling solvents such as toluene and dimethylformamide as reaction solvent, which subsequently require high reaction temperatures, and this in turn leads to tedious workup procedures. In view of these drawbacks, there is a need to develop an industrially viable commercial process for the preparation of lurasidone and its intermediates; which is simple, efficient and cost-effective process and provides the desired compounds in improved yield and purity.
Inventors of the present invention have developed an improved process that addresses the problems associated with the processes reported in the prior art. The process of the present invention does not involve use of any toxic and/or costly solvents. Moreover, the process does not require additional purification steps and critical workup procedure. Accordingly, the present invention provides a process for the preparation of lurasidone and its intermediates, which is simple, efficient, cost effective, environmentally friendly and commercially scalable for large scale operations.

Scheme-II

Scheme-Ill
EXAMPLES
Example-1: Preparation of (3aR,7aR)-4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2,l’-piperazin]-l’-ium methanesulfonate(the compound II)
Charged 150.0 mL (3v) of isopropyl alcohol (IPA) in a flask followed by the addition of the compound-Ill (50.0 g) , 3-(l-Piperazinyl)-l, 2-Benzisothiazole (32.84 g), sodium carbonate granular (10.79 g) and water 50 mL (lv). The reaction mixture was heated at a temperature of 82-85 °C for 24 to 25 h. Cooled the reaction mixture to room temperature, filtered on Buchner funnel and the filtrate was collected.
The filtrate was evaporated under vacuum at 55-65°C till visible solid appears in the reaction mass. The solid was stirred in 75 mL of toluene at room temperature and the solid was filtered. The wet cake was transferred to a flask and added 125 mL of acetone to it; followed by stirring at room temperature. The resulting solid was filtered to yield the pure title compound (II).
Yield: 63.4 g (90 %)
Purity (by HPLC): 99.79 %
Unreacted compound-IV as impurity in 0.05 % .
Example-2: Preparation of Lurasidone free base.
Charged 150.0 mL of Ν,Ν-dimethylformamide (DMF) in a flask followed by the addition of 50.0 g of the compound-II (as obtained in the above example-1), 19.5 g (3aR,4S,7R,7aS)-4,7-methano-lH-isoindole-l,3(2H)-dione and 19.5 g of potassium carbonate. The reaction mixture was heated at a temperature of about 125 °C for 24 h. The reaction mixture was cooled to room temperature and 400 mL of water was added to it. The reaction mixture was stirred, and the precipitated product was filtered. The wet cake was washed with IPA and Lurasidone free base is obtained as the pure product. [Yield: 46.52 g (80 %)]
Example-3: Purification of Lurasidone hydrochloride.
Charged water (200 ml) and IPA (200 ml) in flask followed by the addition of Lurasidone hydrochloride (50 gm, residual acetone: 5769 ppm). The reaction mixture was heated at a temperature of 75-80 °C for about 30 min. The reaction mixture was cooled to 20-30 °C and stirred for about 2 hours. The precipitated solid was filtered and isolated as pure Lurasidone hydrochloride (residual acetone: 2 ppm)
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India
///////////////WO 2016110798, Piramal Enterprises Ltd, New Patent, Lurasidone
Filed under: PATENT, PATENTS Tagged: LURASIDONE, NEW PATENT, Piramal Enterprises Ltd, WO 2016110798







