

 .
.
Picture credit….Bethany Halford
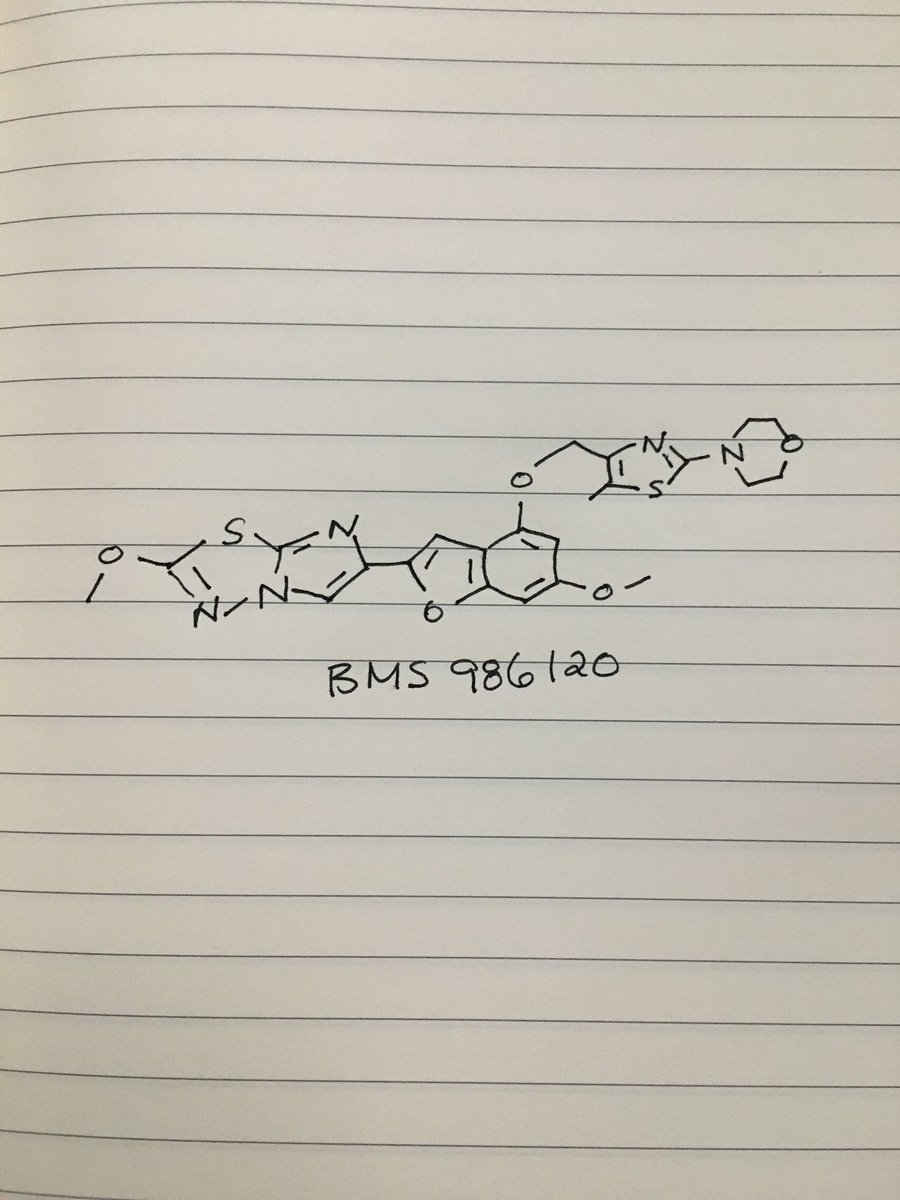
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound


PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\


IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
Filed under: PHASE 1, PHASE1 Tagged: 1478712-37-6, BMS 986120, Bristol-Myers Squibb, Imidazoles, PHASE 1, Small molecules, Thiadiazoles







