
Vismodegib
2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide
Vismodegib; 879085-55-9; GDC-0449; 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide; Erivedge; HhAntag691; CUR-691
GDC-449
Hh-Antag691
HhAntag
R-3616
RG-3616
421.29706 g/mol
LAUNCHED 2012
Vismodegib is a Hedgehog Pathway Inhibitor. The mechanism of action of vismodegib is as a Smoothened Receptor Antagonist.
Hedgehog Antagonist GDC-0449 is an orally bioavailable small molecule with potential antineoplastic activity. Hedgehog antagonist GDC-0449 targets the Hedgehog signaling pathway, blocking the activities of the Hedgehog-ligand cell surface receptors PTCH and/or SMO and suppressing Hedgehog signaling. The Hedgehog signaling pathway plays an important role in tissue growth and repair; aberrant constitutive activation of Hedgehog pathway signaling and uncontrolled cellular proliferation may be associated with mutations in the Hedgehog-ligand cell surface receptors PTCH and SMO.
NMR from net

Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
![]()
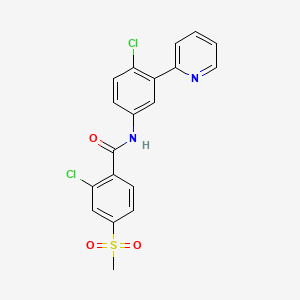
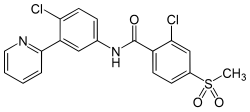
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:

Vismodegib (trade name Erivedge) is a drug for the treatment of basal-cell carcinoma (BCC). The approval of vismodegib on January 30, 2012, represents the first Hedgehog signaling pathway targeting agent to gain U.S. Food and Drug Administration (FDA) approval.[1] The drug is also undergoing clinical trials for metastatic colorectal cancer, small-cell lung cancer, advanced stomach cancer, pancreatic cancer, medulloblastoma and chondrosarcoma as of June 2011.[2] The drug was developed by thebiotechnology/pharmaceutical company Genentech, which is headquartered at South San Francisco, California, USA.
Indication
Vismodegib is indicated for patients with basal cell carcinoma (BCC) which has metastasized to other parts of the body, relapsed after surgery, or cannot be treated with surgery or radiation.[3] [4]
Mechanism of action
The substance acts as a cyclopamine-competitive antagonist of the smoothened receptor (SMO) which is part of the hedgehog signaling pathway.[2] SMO inhibition causes the transcription factors GLI1 and GLI2 to remain inactive, which prevents the expression of tumor mediating genes within the hedgehog pathway.[5] This pathway is pathogenetically relevant in more than 90% of basal-cell carcinomas.[6]
PAPER
Bioorg Med Chem Lett 2009, 19(19): 5576
http://www.sciencedirect.com/science/article/pii/S0960894X10012709

Figure 1.
Schematic for the discovery of 2 (GDC-0449) from 1, and the inspiration for further analogs 3 and 4
CN 103910671
http://www.google.com/patents/CN103910671A?cl=en
In embryonic development, Hedgehog signaling in cell differentiation, tissue development and organogenesis play an important role. In the adult body, Hedgehog signaling pathway is mainly in slumber, but when abnormal tissue growth and self-healing, Hedgehog pathway may be activated. With the in-depth study of the tumor, the presence of numerous evidence of abnormal tumor occurrence and the close relationship between Hedgehog signaling pathway, such as sporadic basal cell carcinoma, medulloblastoma, small cell lung cancer and gastrointestinal cancer and other diseases, therefore Hedgehog signaling pathway targeted anti-cancer therapy inhibitors become hot.
Vismodegib chemical name 2_ chlorine -N_ (4_ chlorine _3_ (_2_ pyridyl) phenyl) _4_ (methylsulfonyl) benzamide, is by Roche’s Genentech (Genentech) Hedgehog pathway inhibitors developed, and can be inhibited by binding seven transmembrane protein Smoothened (Smo), thereby preventing signal transduction. Vismodegib capsule in January 2012 I was approved and listed by the US Food and Drug Administration, under the trade name Erivedge, for the treatment of adults with the most common type of skin cancer – basal cell carcinoma. This medicine is not intended for surgery or radiotherapy of cancer and basal cell skin cancer locally advanced patients have been transferred. This was the first drug approved for the treatment of basal cell carcinoma.

W02006028958 Vismodegib disclose the following synthesis route:
Route One Negishi coupling reactions

wherein, X1 is chloro, bromo or iodo; X2 is bromo, iodo or tosylate. The route to the 2-halo-pyridine as starting material an organic zinc compound, and then prepared by Negishi coupling reaction to give 2- (2-chloro-5-nitrophenyl) pyridine. 2- (2-chloro-5-nitrophenyl) pyridine in turn through a reduction reaction with acylation reaction, to give the final product Vismodegib. The key coupling step of the route using an organic zinc reagent required to react under strict anhydrous, anaerobic conditions.
The second route Suzuki coupling reaction [0010]

wherein, X2 is bromo, iodo or tosylate. The route from 3-halo-4-chloro-nitrobenzene as raw material, and 2-chloro-5-nitrophenyl boronic acid pinacol ester, and then reacted with a 2-halo-pyridine was prepared to give 2- (2-chloro 5-nitrophenyl) pyridine. 2- (2-chloro-5-nitrophenyl) pyridine then after reduction and acylation reaction, to give the final product Vismodegib. The key coupling step of the route using the Suzuki coupling reaction, organic boron reagent price to use expensive, high production costs.
The route three Suzuki coupling reaction

wherein, X2 is bromo, iodo or tosylate. Similar to the second route, the route is still critical coupling step using a Suzuki coupling reaction, the same need to use expensive organic boron reagents, higher production costs.
route four Stille coupling reaction

The route to 2-p-toluenesulfonyl pyridine as starting material, is reacted with an organotin reagent, prepared to give pyridin-2-yl trimethyltin, then by Stille coupling reaction, was prepared to give 2- (2-chloro – 5- nitrophenyl) pyridine, followed by reduction reaction, acylation prepared to give Vismodegib. The key step of the route using the Stille coupling reaction, this step need to use expensive and toxic organotin reagents, and the need to carry out the reaction under strict anhydrous, anaerobic conditions.
A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising: a compound of formula III was prepared as a compound of Formula II;

Then, the compound of formula II with a compound of formula I, to give 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide;

Wherein, R1 is halogen or hydroxy, preferably chlorine, or a hydroxyl group.
2. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:

Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
3. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:

Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
Example 1: N–oxo-2- (2-chloro-5-nitrophenyl) pyridine

[0108] To a 100mL three-necked flask were added 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol potassium carbonate, 0.05mmol tri-butyl acetate button and 0.15mmol phosphorus tetrafluoroborate salt, 40ml of toluene, IS gas exchange three times, under argon at reflux for 2 days, then the reaction mixture was poured into 100mL of ethyl acetate, filtered, and the filtrate was washed with saturated brine, dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50), fractions were collected and the solvent was distilled off under reduced pressure to give a pale yellow solid, yield 60%.
1HMffi (500Hz, DMS0_d6): 8.35 (m, 3H), 7.90 (d, 1Η), 7.62 (q, 1Η), 7.55 (m, 1Η), 7.48 (m, 1Η);
MS: 251.1,253.1 ([Μ + Η] +).
2 Example: Ν–oxo-2- (2-chloro-5-nitrophenyl) pyridine

To a 100mL three-necked flask 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol of potassium carbonate, 0.05mmol iodide and 0.1Ommoll, 10- Fei Luo Jie morpholine, 40ml of xylene, an argon gas exchange three times, under argon at reflux for 2 days, cooled to room temperature and then the reaction system was poured into 100mL methylene chloride, filtered and the filtrate washed with saturated brine, dried, filtered, The filtrate solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50) to give a pale yellow solid, yield 42%. .
3 Example: 2- (2-chloro-5-nitrophenyl) pyridine

After 3.0mmol N- oxo added to 100mL of Lord vial _2_ (2_ chloro _5_ nitrophenyl) pyrazole 唳, 15mmol phosphorus trichloride and 30ml of chloroform was heated at reflux for 12h, the reaction It was poured into 100mL of water and extracted with ethyl acetate (50ml X 2), and the combined organic phase was dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: petroleum ether / ethyl acetate = 20/1) , fractions were collected, the solvent was distilled off under reduced pressure to give a white solid, yield 95%.
1Hnmr (SooHzJDCI3): 8.78 (d, 1H), 8.51 (d, 1H), 8.20 (m, 1H), 7.85 (m, 1H), 7.72 (d, 1H), 7.65 (d, 1H), 7.40 (m, 1H);
MS: 235.1,237.1 ([M + H] +).
4 Example 2: Preparation 4_ chlorine _3_ (topiramate 唳 _2_ yl) aniline

To a vial was added 100mL of Lord 20mmol2- (2- chloro-5-nitrophenyl) pyridine 唳, 50ml of acetic acid, heated to 80 ° C and stirred, and then slowly added IOOmmol iron, reaction 0.5h The reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X 3), the combined organic phases, the organic phase was washed with saturated sodium carbonate solution (50ml X 3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a pale yellow solid, yield 75%.
1HMflUSOOHz, DMS0_d6): 8.63 (m, 1H), 7.84 (m, 1H), 7.56 (d, 1H), 7.37 (m, 1H),
7.13 (d, 1H), 6.76 (d, 1H), 6.61 (q, 1H), 5.32 (s, 2H);
MS: 205.1,207.1 ([M + H] +).
5 Example: 4-chloro-3- (pyridin 唳-2-yl) aniline

to 100mL of God-shaped flask 20mmol2_ (2_ chlorine _5_ nitrophenyl) pyridine Jie set, 50ml of methanol, Ig activated carbon, 2mmol FeOOH and 60mmol85% of hydrazine hydrate, heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, the organic phase was washed with saturated sodium bicarbonate solution (20ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product was recrystallized from n-propanol to give a pale yellow solid, yield 96%.
6 Example 2: Preparation 4_-chloro-3- (2-yl) aniline

20mmol N- oxo added to 100mL eggplant-shaped flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of acetic acid, heated to 80 ° C and stirred, and then iron powder was slowly added IOOmmol After 0.5h the reaction the reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X3), the combined organic phases were washed with saturated sodium carbonate solution (50ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propanol recrystallized to give a white solid, yield 70%.
Preparation 7.Α ~ chlorine -3_ (topiramate 唳 2-yl) aniline [0130] Example

20mmol N- oxo added to 100mL eggplant type flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of methanol, Ig active carbon, 2mmol FeOOH 60mmol85% hydrazine hydrate and heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, washed with saturated aqueous sodium bicarbonate solution, the organic phase (20mlX3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a white solid, yield 82%.
Vismodegib Preparation: 8 Example

In the Lord 50ml vial, the 1.50mmol2- chloro-4-methanesulfonyl-chloride in 15ml of dry tetrahydrofuran, cooled to ice bath O ~ 10 ° C, a solution of 4-chloro-3 – (pyridin-2-yl) aniline in anhydrous tetrahydrofuran (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction system was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
1HNMR (500Hz, DMS0_d6): 10.90 (s, 1H), 8.70 (d, 1H), 8.12 (d, 1H), 8.01 (t, 2H), 7.92 (m, 2H), 7.74 (q, 1H ), 7.69 (d, 1H), 7.58 (d, 1H), 7.44 (m, 1H), 3.34 (s, 3H).
MS: 421.1,423.1 ([M + H] +).
Vismodegib Preparation: 9 Example

In 50ml vial of God, will 1.50mmol2_ chlorine _4_ methylsulfonyl benzoic acid, 1.47mmol4_ chlorine _3_ (batch 唳 2-yl) aniline and triethylamine were dissolved in 25ml 2.5mmol anhydrous tetrahydrofuran in an ice bath to cool to O ~ 10 ° C, was added in portions N, N ‘- dicyclohexyl carbodiimide (DCC) 1.50mmol, After the addition, the reaction at room temperature 6h, after the reaction, white solid was removed by filtration, the filtrate was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 84%.
Vismodegib Preparation: 10 [0141] Example

In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
PATENT
CN 103910672
http://www.google.com/patents/CN103910672A?cl=en
Vismodegib PreparatioN

In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
PATENT
WO2006028958
https://www.google.co.in/patents/WO2006028958A2?cl=en
Example 1 General Procedure
Compounds of examples 2-51 were prepared according to the following general procedures.
A: Suzuki Coupling Procedure

2 M aq. Potassium carbonate (5.0 eq) and 4:1 toluene :ethanol mixture (2.5 mL) were added to a microwave vial charged with the appropriate boronate ester (2.6 eq), aryl halide (0.35 mmol, 1.0 eq), and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 160 0C for ten minutes. The solution was poured onto 2 M aq. Sodium hydroxide (20 mL), extracted with ethyl acetate (2 x 20 mL), dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
B: Negishi Coupling Procedure

X = I or Br R = H, 3-Me, 4-Me5 5-Me, 6-Me
Aryl zinc bromide (0.5 M in THF, 2.5 eq) was added to an oven-dried microwave vial charged with the appropriate aryl halide (1.0 eq) and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 140 0C for 10 minutes. The crude reaction mixture was concentrated and purified by chromatography on silica gel (conditions given below) to afford the desired product.
C: Iron Reduction of Aryl Nitro Group

R = I or pyridin-2-yl
The appropriate nitro aryl (1 mmol, 1 eq) in AcOH/EtOH (1:1, 0.42 M) was added slowly to a solution of Iron powder (6.0 eq) in AcOH/EtOH (1:2, 2 M) at 60 °C. The solution was stirred at 70 0C for 30-60 minutes. The reaction mixture was cooled to 23 0C, filtered through celite, washed with ethyl acetate, and concentrated. The oily residue was dissolved in ethyl acetate (30 mL), washed with saturated aq. NaHCO3 (2 x 15 rnL) and water (2 x 10 niL), dried (MgSO4), and concentrated. The oily residue was used with out further purification.
D: Amide Bond Formation

R = I or pyridin-2-yI
Acid chloride (1.05-1.1 eq) was added to a solution of aniline (1.0 eq) and TEA (1.1-1.5 eq) in methylene chloride at the indicated temperature. The solution was stirred for 0.5-3 hours, poured onto saturated aq. NaHCO3, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
E: EDC Amide Bond Formation

R = I or pyridin-2-yl
Carboxylic acid (1.1 eq) was added to a solution of aniline (1.0 eq) and EDC (1.4 eq) in methylene chloride (0.7 M in aniline). The solution was stirred at 23 0C for 2 hours, poured onto a 1 :1 mixture of saturated aq. NH4Cl and water, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product. F: addition of amines to 2-chloropyridine

NHRR’ = ethanolamine, analine, benzylamine, 2-methylpropylamine, N-methylpiperazine, morpholine, 2-morpholinoethylamine
Primary or secondary amine (5 eq) in either BuOH or a mixture of BuOH/ethylene gylcol was heated to 170 to 220 0C for 20 min in a sealed tube. The BuOH was removed under reduced pressure. In cases where ethylene glycol was used, the reaction was diluted with water, and the product was extracted into ethyl acetate, dried (MgSO^, and concentrated. The crude residue was purified by reverse phase HPLC to afford the desired product.
G: Amide bond coupling with HATU
HATU, DIPEA, DMF NaOH or NaHCO3

ethyl acetate extraction

Aniline (1.0 eq) was added to a mixture of carboxylic acid (1.1 eq), HATU (1.1 eq) and DIPEA (2 eq) in DMF (0.1 – 0.2 M). After stirring overnight, the reaction mixture was diluted with 0.1 N sodium hydroxide or saturated NaHCθ3, extracted into ethyl acetate and the combined organic layers were washed with brine. The organic layer was dried (MgSO4), concentrated and the crude mixture was purified by reverse phase HPLC. H: Preparation of sulfonamide benzoic acids

Chlororsulfonylbenzoic acid (1.0 eq) was added to a solution of amine (1.1 eq) in 10-20% DEPEA/methanol (1 M) at 4 0C. After 1 h, the reaction mixture was concentrated, and the crude residue was purified by reverse phase HPLC.
I : Stannylation of 2-pyridyl triflates

A solution of tetrakis-triphenylphosphinepalladium (0.04 eq.) in toluene (1 mL) was added to degassed solution of aryltriflate (1 eq), bis-trialkyltin (1.05 eq), and lithium chloride (3 eq) in dioxane. Heated to reflux for 2 hours, cooled to 23 0C, diluted with ethyl acetate, washed with 10% NH4θH(aq) and brine, dried (MgSO4) and concentrated. The crude material was used without further purification.
J: Stannylation of substituted pyridines

ιMmβco3 n-Butyl lithium (6 eq, 2.5 M in hexanes) was added dropwise to a solution of dimethylaminoethanol (3 eq) in hexane at 0 0C. The solution was stirred at 0 0C for thirty minutes before dropwise addition of the substituted pyridine (1 eq). The solution was stirred at 0 0C for an additional hour, then cooled to -78 0C. A solution of trialkyltin in hexane was added dropwise. The solution was stirred at -78 0C for thirty minutes, warmed to 0 0C, quenched with water, extracted twice with ether, dried (MgSO4), and concentrated. K: Stille Coupling

Palladium catalyst (0.02 eq) was added to a degassed solution of aryliodide (1 eq), arylstannane (2 eq), and triphenylphosphine (0.16 eq) in NMP. Heated in the microwave to 130 0C for 15 minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC>4), concentrated and purified by silica gel chromatography.
L: Synthesis of alky lethers

A solution of hydroxypyridine (1 eq), alkyliodide (excess), and cesium carbonate in NMP was heated in the microwave to 1000C for ten minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC^), concentrated and purified by silica gel chromatography.
M: Methyl Ester Saponification

The methyl ester (leq) was hydrolyzed with LiOH (2eq) in 50/50 THF/water mix. Upon completion of the reaction the THF was evaporated under reduced pressure and the solution is acidified with HCl to pH 2. The resultant solid was filtered and dried to give the pure acid.
N: Bromination in the presence of a free acid functionality
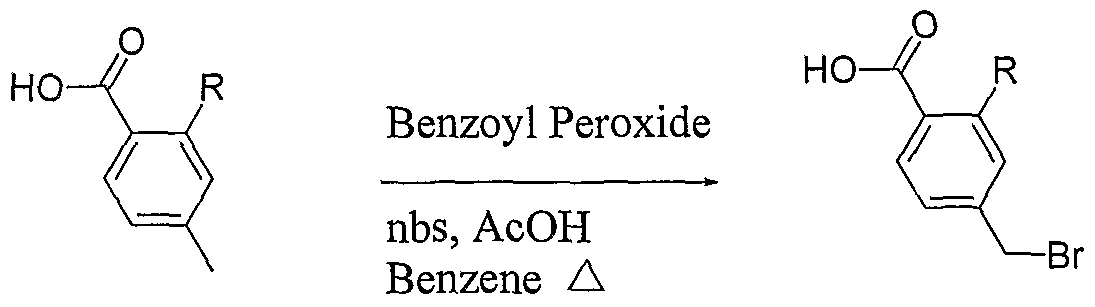
The paramethylbenzoic acid (leq) was combined with Benzoyl Peroxide (O.leq) and N- Bromosuccinimde (0.9eq) in a solution of 5%AcOH in Benzene and heated in the microwave at 120°C for 5-15minutes. The product was separated from the starting material and di-bromo product via ISCO flash chromatography with an ethyl acetate (with 1% AcOH) and hexanes solvent system.
O: Sodium Methanesulfinate displacement of Bromine

To the bromine starting material (leq) was added sodium methanesulfinate (2eq) in DMF and heated to 120°C in the microwave for 5 minutes. Alternatively, the reaction was heated to 60°C in an oil bath for several hours until completed. Reaction mixture was concentrated under reduced pressure and extracted in ethyl acetate and water. The organic layer was dried over Magnesium Sulfate, filtered and concentrated in vacuo to yield generic methylsulfone.
P: Amine displacement of Bromine

To the bromo starting material (leq) was added appropriate amine (3eq) in either DMSO or BuOH and stirred at room temperature until complete. For less nucleophilic amines or anilines, the reactions were forced to completion using microwave conditions ranging from 150°-170°C for 15 minutes. Crude reactions were concentrated to dryness and either extracted with ethyl acetate and saturated bicarbonate if the reaction resulted in an intermediate or purified via HPLC if the reaction resulted in a final product.
Q: Thiol displacement of halogen

The paramethylbromo benzoate (leq) was treated with Potassium (or Cesium) Carbonate (1.5eq) and appropriate thiol derivative (l,leq) in DMF (or CH3CN) and stirred overnight at room temperature. The DMF was evaporated in vacuo and the reaction was extracted with ethyl acetate and water. The organic layer was dried over Magnesium Sulfate , filtered and concentrated to yield the thiol or derivatized thiol compound.
R: Oxone Oxidation
oxone 2:1 MeOHTH2O


Derivatized thiol (leq) was dissolved in MeOH while Oxone (2eq) was seperately dissolved in half the amount of water. Once all the oxone was dissolved, the solution was added to the thiol in MeOH solution at once and stirred until complete. The MeOH was evaporated in vacuo and the remaining water was extracted twice with Ethyl Acetate. The organic layer was dried over Magnesium Sulfate and concentrated to yield the sulfone.
S: Thio lysis of epoxides at alumina surfaces

A mixture of epoxides (1.0 eq), thiophenol (1.5 eq) and neutral aluminum oxide (~70 eq) in diethyl ether was stirred for 3 h at room temperature while being monitored by TLC. The reaction mixture was filtered through Celite, washed with ethyl acetate and concentrated. Purified by silica gel chromatography (0-40% ethyl acetate/hexane) to yield β -hydroxysulfide product.
T: Conversion of nitrile group to carboxylic acid

R
A solution of benzonitrile (1.0 eq) and sodium hydroxide (2.0 eq) in H2O was heated to 120 ° C for 2h. The reaction mixture was cooled to room temperature and acidified with HCl to pH 2. The resulting solid was filtered to afford the pure acid product.
U. Alkylation of phenols

The phenol was dissolved in DMF (1.0 ml). Cesium carbonate (1.0 eq.) and an alkyl bromide or alkyl iodide (1.0 to 2.0 eq.) were added, and the reaction was stirred at room temperature for 18 hrs or 5O0C for 1 to 24 hours. The reaction was quenched in water, and extracted with ethyl acetate twice. The organic extracts were washed with water once, brine once, dried with MgSC>4, and evaporated to a crude oil which was purified on reverse phase HPLC.
V. Amide bond formation with an acid chloride and an aniline

The aniline was dissolved in THF (1.5 ml) and dichloromethane (1.5 ml). MP-Carbonate (1.5 eq.) and an acid chloride (1.1 eq.) were added, and the solution was stirred at room temperature for 18 hours. The reaction was diluted with methanol and dichloromethane, and filtered to remove the MP-Carbonate. The mother liquors were evaporated to a solid and purified by reverse phase HPLC.
W. Amidine formation from an imidate

A solution of freshly formed imidate in methanol was treated with a primary or secondary amine (1.5 eq.) at room temperature for 18 hours. The methanol was removed on a rotary evaporator and the residue purified by reverse phase HPLC.
Example 37 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide

Procedure G was used to couple 4-chloro-3-(pyridin-2-yl)aniline (50 mg) and 2-chloro-4- methylsulfonylbenzoic acid to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4- (methylsulfonyl)benzamide. MS (Ql) 421.0 (M)+. The product was then dissolved in 1 Ν HCI solution followed by freebasing with 0.5 Ν NaOH solution (pH to 11). The resulting precipitate was filtered and vacuum-dry.
Procedure D may also be used to couple 4-chloro-3-(pyridin-2-yl)aniline and 2-chloro-4- (methylsulfonyl)benzoyl chloride to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-
(methylsulfonyl)benzamide which is collected by suction filtration and the HCl salt is washed with
Et2O (or alternatively with MTBE). This material is freebased using EtOAc/aq NaHCO3 and the organics are dried and concentrated to the solid freebase. This material is then crystallized from acetone :EtOAc (80:20, approx lOmL/g) which is then finally recrystallized from hot slurry of iPrOAc. 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide HCl salt may also be dissolved in distilled water followed by freebasing with 0.5 N NaOH solution (pH to 11) and filtering and vacuum drying the precipitate.
Patent
WO 2016020324, BASF AG, vismodegib , new patent
WO2016020324, MULTI-COMPONENT CRYSTALS OF VISMODEGIB AND SELECTED CO-CRYSTAL FORMERS OR SOLVENTS
BASF SE [DE/DE]; 67056 Ludwigshafen (DE)
VIERTELHAUS, Martin; (DE).
CHIODO, Tiziana; (DE).
SALVADOR, Beate; (DE).
VOSSEN, Marcus; (DE).
HAFNER, Andreas; (CH).
HINTERMANN, Tobias; (CH).
WEISHAAR, Walter; (DE).
HELLMANN, Rolf; (DE)
![]()
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 and a second compound selected from the group consisting of co-crystal formers and sol-vents. The invention is further related to pharmaceutical compositions comprising such multi-component crystals. Furthermore, the invention relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease.
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:
formula 1
Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 (cf. above) and a second compound selected from the group consisting of co-crystal formers and solvents.
The invention is further related to pharmaceutical compositions comprising said multi-component crystals. Furthermore, the invention also relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease. Further details as well as further aspects of the present invention will be described herein below.
Vismodegib is a BCS class II compound with a high permeability but a low solubility where enhanced solubility or dissolution rates can lead to a significant advantage in respect to bio-availability.
Vismodegib is known to exist as crystalline free base. Salts of Vismodegib are men-tioned in US 7,888,364 B2 but not specified. In particular, the HCI salt is mentioned as intermediate but not characterized. Co-crystals or solvates are not reported at all.
The solubility of Vismodegib is reported to be 0.1 μg/mL at pH 7 and 0.99 mg/mL at pH 1 for Erivedge®. The absolute bio-availability after single dose is reported to be 31.8 % and the ex-posure is not linear at single doses higher than 270 mg. Erivedge® capsules do not have a food label. The estimated elimination half-life (t1/2) after continuous once-daily dosing is 4 days and 12 days after a single dose treatment (Highlights of Prescribing Information: ERIVEDGE® (vismodegib) capsule for oral use; Revised: 01/2012).
The discovery and preparation of new co-crystals or solvates offer an opportunity to improve the performance profile of a pharmaceutical product. It widens the reservoir of techniques/materials that a formulation scientist can use for designing a new dosage form of an active pharmaceutical ingredient (API) with improved characteristics. One of the most important characteristics of an API such as Vismodegib is the bio-availability which is often determined by the aqueous solubility.
A compound like Vismodegib may give rise to a variety of crystalline forms having dis-tinct crystal structures and physical characteristics like melting point, X-ray diffraction pattern, infrared spectrum, Raman spectrum and solid state NMR spectrum. One crystalline form may give rise to thermal behavior different from that of another crystalline form. Thermal behavior can be measured in the laboratory by such techniques as capillary melting point, thermogravimetry (TG), and differential scanning calorimetry (DSC) as well as content of sol-vent in the crystalline form, which have been used to distinguish polymorphic forms.
Multi-component crystals comprising Vismodegib and selected co-crystal formers or solvents may improve the dissolution kinetic profile and allow to control the hygrosco-picity of Vismodegib.
Therefore, there is a need for multi-component crystals comprising Vismodegib that avoid the above disadvantages. In particular, it is an object of the present invention to provide multi-component crystals of Vismodegib with optimized manufacture, formula-tion, stability and/or biological efficacy
.
Example 1 :
314 mg Vismodegib and 86 mg maleic acid are suspended in toluene saturated with maleic acid for 2 d, filtered and dried.
TG data shows a mass loss of about 2.3 wt % between 100 and 1 18 °C which is attributed to rest solvent. DSC data shows a single endothermal peak with an onset of about 1 15 °C (99 J/g).
H-NMR spectroscopy indicates a molar ratio of Vismodegib to maleic acid of about 1 :1 .3. However single crystal X-ray data confirms a ratio of 1 :2 (Table 1 ).


update……………
Vismodegib Synthesis
WO2009126863A2: also see Ref. 1. It all started from here.

Identification:

| 1H NMR (Estimated) for Vismodegib |
Experimental: 1H NMR (400MHz, CDCl3) δ (ppm): 9.58 (bs, 1H), 8.43 (d, J = 4.7Hz, 1H), 8.03 (dd, J = 2.6, 8.7Hz, 1H), 7.90 (d, J = 1.6Hz, 1H), 7.67-7.78 (m, 4H), 7.60 (d, J = 8.0Hz, 1H), 7. 51 (d, J = 8.8Hz, 1H), 7.23-7.24 (m, 1H), 3.01 (s, 3H).
References
- “Vismodegib, First Hedgehog Inhibitor, Approved for BCC Patients”.
- “Molecule of the Month”. June 2011.
- “FDA approves Erivedge (vismodegib) capsule, the first medicine for adults with advanced basal cell carcinoma”.
- Lacroix, Marc (2014). Targeted Therapies in Cancer. Hauppauge , NY: Nova Sciences Publishers. ISBN 978-1-63321-687-7.
- “Vismodegib (GDC-0449) Smoothened Inhibitor – BioOncology”.
- H. Spreitzer (4 July 2011). “Neue Wirkstoffe – Vismodegib”. Österreichische Apothekerzeitung (in German) (14/2011): 10.
- FDA Professional Drug Information
External links
- Erivedge® (vismodegib), a prescription oral medica on approved for advanced basal cell carcinoma treatment
- Efficacy and Safety of Vismodegib
- Food and Drug Administration (FDA) approved vismodegib
PatentSubmittedGranted
Pyridyl inhibitors of hedgehog signalling [US7888364]2006-03-232011-02-15
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2009281089]2009-11-12
ANTI-HEDGEHOG ANTIBODIES [US8030454]2010-01-072011-10-04
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2011092461]2011-04-21
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2012094980]2011-10-142012-04-19
COMBINATION THERAPY WITH NANOPARTICLE COMPOSITIONS OF TAXANE AND HEDGEHOG INHIBITORS [US2013045240]2010-08-252013-02-21
COMBINATION THERAPY WITH NANOPARTICLE COMPOSITIONS OF TAXANE AND HEDGEHOG INHIBITORS [US2014072630]2013-02-282014-03-13
Acyl guanidine derivatives modulating the hedgehog protein signaling pathway [US8889678]2010-07-192014-11-18
COMBINATION THERAPY [US2012184529]2012-01-032012-07-19
METHOD OF INHIBITING DYRK1B [US2014371251]2014-06-182014-12-18
USE OF SUBSTITUTED HEXITOLS INCLUDING DIANHYDROGALACTITOL AND ANALOGS TO TREAT NEOPLASTIC DISEASE AND CANCER STEM AND CANCER STEM CELLS INCLUDING GLIOBLASTOMA MULTIFORME AND MEDULLOBLASTOMA [US2014377336]2013-01-222014-12-25
SHH Regulation and Methods Thereof [US2012082623]2011-09-302012-04-05
NOVEL 2-PIPERIDIN-1-YL-ACETAMIDE COMPOUNDS FOR USE AS TANKYRASE INHIBITORS [US2015025070]2012-07-132015-01-22
Compositions and Methods for Modulating Neuron Degeneration and Neuron Guidance [US2011065645]2010-09-102011-03-17
SMOOTHENED ANTAGONISM FOR THE TREATMENT OF HEDGEHOG PATHWAY-RELATED DISORDERS [US2014200217]2014-01-242014-07-17
| CN101072755A * | Sep 2, 2005 | Nov 14, 2007 | 遗传技术研究公司 | Pyridyl inhibitors of hedgehog signalling |
| CN102731373A * | Jul 19, 2012 | Oct 17, 2012 | 南京药石药物研发有限公司 | Preparation method of intermediate of antitumor drug GDC-0449 (vismodegib) |
| US20080132698 * | Nov 30, 2006 | Jun 5, 2008 | University Of Ottawa | Use of N-oxide compounds in coupling reactions |
| US20090076266 * | Sep 10, 2008 | Mar 19, 2009 | The University Of Houston System | Copper-catalyzed c-h bond arylation |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | GEORGETTE M. CASTANEDO,等: “Second generation 2-pyridyl biphenyl amide inhibitors of the hedgehog pathway“, 《BIOORGANIC & MEDICINAL CHEMISTRY LETTERS》, vol. 20, 15 September 2010 (2010-09-15), pages 6748 – 6753 |
| 2 | * | 曹萌,等: “Vismodegib 的合成“, 《第十一届全国青年药学工作者最新科研成果交流会论文集》, 21 June 2012 (2012-06-21) |
| 3 | * | 耿一丁: “Vismodegib“, 《中国药物化学杂志》, vol. 22, no. 3, 20 June 2012 (2012-06-20) |
| 4 | * | 邢其毅,等: “《基础有机化学》”, 31 December 2005, article “201310019450.0“, pages: 896-897 |
 |
|
|
|
| Systematic (IUPAC) name | |
|---|---|
|
2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide
|
|
| Clinical data | |
| Trade names | Erivedge |
| AHFS/Drugs.com | monograph |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 31.8% |
| Protein binding | >99% |
| Metabolism | <2% metabolised byCYP2C9, CYP3A4, CYP3A5 |
| Biological half-life | 4 days (continuous use), 12 days (single dose) |
| Excretion | Faeces (82%), urine (4.4%) |
| Identifiers | |
| CAS Number | 879085-55-9 |
| ATC code | L01XX43 |
| PubChem | CID 24776445 |
| IUPHAR/BPS | 6975 |
| DrugBank | DB08828 |
| ChemSpider | 23337846 |
| UNII | 25X868M3DS |
| ChEBI | CHEBI:66903  |
| ChEMBL | CHEMBL473417 |
| Synonyms | GDC-0449, RG-3616 |
| Chemical data | |
| Formula | C19H14Cl2N2O3S |
| Molar mass | 421.30 g/mol |
SEE…http://apisynthesisint.blogspot.in/2016/02/vismodegib.html
/////
CS(=O)(=O)C1=CC(=C(C=C1)C(=O)NC2=CC(=C(C=C2)Cl)C3=CC=CC=N3)Cl
CS(=O)(=O)C1=CC(=C(C=C1)C(=O)NC2=CC(=C(C=C2)Cl)C3=CC=CC=N3)Cl
Filed under: FDA 2012, Uncategorized Tagged: Erivedge, fda 2012, vismodegib







