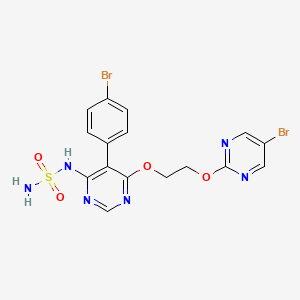
Aprocitentan, sold under the brand name Tryvio, is a medication used to treat hypertension (high blood pressure).[1] It is developed by Idorsia.[2] It is taken by mouth.[1]
Aprocitentan is indicated for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adults who are not adequately controlled on other medications.[1]
Data from animal reproductive toxicity studies with other endothelin-receptor agonists indicate that use is contraindicated in pregnant women.[1]
Mechanism of action
Aprocitentan is an endothelin receptor agonist that inhibits the protein endothelin-1 from binding to endothelin A and endothelin B receptors.[1][4] Endothelin-1 mediates various adverse effects via its receptors, such as inflammation, cell proliferation, fibrosis, and vasoconstriction.[1]
Society and culture
Economics
Aprocitentan is developed by Idorsia, which sold it to Janssen and purchased the rights back in 2023, for US$343 million.[6]
Legal status
Aprocitentan was approved for medical use in the United States in March 2024.[1]
In April 2024, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Jeraygo, intended for the treatment of resistant hypertension in adults.[7] The applicant for this medicinal product is Idorsia Pharmaceuticals Deutschland GmbH.[7]
The following example was prepared according to the procedures described below. All compounds were characterized by 1H-NMR (300 MHz) and occasionally by 13C-NMR (75 MHz) (Varian Oxford, 300 MHz; chemical shifts are given in ppm relative to the solvent used; multiplicities: s=singlet, d=doublet, t=triplet; m=multiplet), by LC-MS (Finnigan Navigator with HP 1100 Binary Pump and DAD, column: 4.6×50 mm, Develosil RP Aqueous, 5 μm, 120 A, gradient: 5-95% acetonitrile in water, 1 min, with 0.04% trifluoroacetic acid, flow: 4.5 ml/min), t R is given in min; by TLC (TLC-plates from Merck, Silica gel 60 F 254) and occasionally by melting point.
Preparation A: Benzylsulfamide Potassium Salt
A.i. Benzylsulfamide
Chlorosulfonylisocyanate (14.14 g) was dissolved in DCM (50 mL) and cooled to 0° C. A solution of t-BuOH (9.6 mL) in DCM (50 mL) was added within 30 min. Stirring was continued for additional 30 min at rt. The solution thus obtained was then added at 0° C. within 1 h to a solution of benzylamine (10.7 g) and TEA (15.32 mL) in DCM (200 mL). Stirring is continued for 10 h at rt. The mixture was concentrated in vacuo, taken up in EA (500 mL) and washed with water (2×40 mL) and brine (30 mL), dried over MgSO 4, filtered. The filtrate was concentrated in vacuo and the crude material was crystallized from EA and dried under HV to give N-benzyl-N′-tert-butoxycarbonyl sulfamide (13.68 g).
This material was dissolved in dioxane (20 ml) and 4 M HCl in dioxane (120 mL) was added within 1 h at rt. The mixture was stirred for 8 h before the solvent was evaporated and the residue dried under HV to give benzylsulfamide as an off-white powder (9.47 g).
To a solution of benzylsulfamide (17.98 g) in McOH (300 mL) was carefully added potassium tert-butylate (10.8 g). The mixture was stirred at rt for 15 min before the solvent was evaporated. The remaining residue was dried under HV to give benzylsulfamide potassium salt as an off-white powder (21.73 g).
To a solution of 4-bromophenylacetic acid (50 g) in methanol (250 ml) was added dropwise thionyl chloride (34.2 mL) while the temperature of the reaction mixture was kept at 0-5° C. Upon complete addition cooling was stopped and the mixture was allowed to warm to rt. Stirring was continued for 75 min before the solvent was removed in vacuo. The yellow oil was dissolved in benzene and again concentrated. The residue was dissolved in EA, washed with water, brine, 2 N aq. Na 2CO 3, and again brine. The org. extract was dried over MgSO 4, filtered, concentrated and dried under HV at 85° C. for 30 min to give the expected product as a yellow oil (52.4 g).
At 40° C., a solution of intermediate B.i (52 g) in THF (100 mL) was carefully added over a period of 40 min to a suspension of NaH (15.6 g) in dry THF (450 mL). Stirring was continued for 70 min without heating and the temperature dropped to 27° C. The evolution of gas stopped before dimethylcarbonate (76.42 mL) was added dropwise while the temperature of the mixture was maintained at 29-31° C. Stirring was continued for 22 h at rt. The mixture was cooled to −10° C. and then carefully neutralized to pH 6-7 with aq. HCl before bulk of the THF was removed in vacuo. The residue was dissolved in EA (700 mL), washed 3 times with 1 N aq. HCl-solution and once with brine, dried over MgSO 4. Most of the EA was evaporated before Hex was added. The product crystallised overnight at 4° C. The crystals were collected, washed with Hex and dried to give the expected product as pale yellow crystals (45.9 g).
A solution of intermediate B.ii (11.73 g) in MeOH (100 mL) was added at 0° C. to a solution of sodium (2.83 g) in MeOH (100 mL). The mixture was stirred for 18 h at rt before formamidine hydrochloride (4.10 g) was added. The suspension was stirred at rt for 4 h. The solvent was removed and the residue was suspended in 10% aq. citric acid (100 mL) and stirred for 10 min. The white precipitate was collected, washed with 10% aq. citric acid, water, evaporated three times from cyclohexane and dried under HV at 40° C. to give 5-(4-bromophenyl)-pyrimidine-4,6-diol as a pale beige powder (9.90 g).
LC-MS: t R=0.62 min, [M+H] +=266.89/268.89 (Br-isotopes).
B.iv. 5-(4-bromo-phenyl)-4,6-dichloro-pyrimidine
To a suspension of 5-(4-bromophenyl)-pyrimidine-4,6-diol (9.90 g) in POCl 3 (130 mL) was carefully added N,N-dimethylaniline (13.5 mL). The mixture is heated to 130° C. for 2 h. The dark brown solution is concentrated in vacuo and the residue was poured into ice/water. The suspension is diluted with 2 N HCl and water and stirred for 20 min. The precipitate that formed is collected and washed with water. The solid material is dissolved in EA, washed with 1 N aq. HCl and brine. The org. phase is dried over MgSO 4 and evaporated. The material is further purified by column chromatography on silica gel eluting with Hex:EA 95:5 to 1:1 followed by crystallisation from Hex/EA at −20° C. to give 4,6-dichloro-5-(4-bromophenyl)-pyrimidine as pale yellow crystals (8.3 g).
A solution of 5-(4-bromophenyl)-4,6-dichloro-pyrimidine (4.00 g, 13.2 mmol) and benzylsulfamide potassium salt (7.38 g, 32.9 mmol) in DMSO (30 mL) was stirred at it for 24 h before being diluted with a 10% aq. citric acid solution (200 mL). The suspension that formed was filtered. The collected solid was washed well with water and dried under HV at 40° C. for 48 h to give the expected product as a white powder (6.15 g).
t-BuOK (18.5 g, 164.5 mmol) was added portionwise to a suspension of intermediate 1.i (7.46 g, 16.4 mmol) in ethylene glycol (50 mL). The mixture became warm and thick and was diluted with DME (75 mL). The mixture was stirred at 95° C. for 24 h before it was cooled to rt, diluted with water (50 mL) and a 10% aq. citric acid solution (250 mL). The milky suspension was extracted with EA (2×300 mL). The combined org. extracts were dried over MgSO 4, filtered and the filtrate was concentrated. The remaining crystalline solid was suspended in MeOH, collected, washed well with MeOH and dried under HV to give the expected product as a white crystalline powder (6.49 g).
To a solution of intermediate 1.ii (6.49 g, 13.5 mmol) in THF (120 mL) was added carefully NaH (1.77 g, 40.6 mmol, 55% dispersion in mineral oil). The mixture was stirred for 10 min before 2-chloro-5-bromo-pyrimidine (3.93 g, 20.3 mmol) was added. The mixture was diluted with DMF (15 mL) and then stirred at rt for 20 min. The mixture was heated to 60° C. and stirred for 3 h before being again cooled to rt. The reaction was quenched with water and 10% aq. citric acid solution (250 mL) and the mixture was extracted with EA (2×300 mL). The org. extracts were washed with water, combined, dried over MgSO 4, filtered and the solvent of the filtrate was evaporated. The crude product was crystallised from MeOH/ether. The crystalline material was collected, washed with additional MeOH/ether and dried under HV to give the expected product as a white powder (6.47 g).
A solution of borontribromide (25.5 mL, 1 M in DCM) was slowly added to a solution of intermediate 1.iii (6.50 g, 10.2 mmol) in chloroform (250 mL). The mixture became turbid and an oily residue separated. The mixture was stirred at rt. Another portion of BBr 3 solution (5 mL) was added after 6, 24, and 33 h. After the last addition of BBr 3, the beige suspension was stirred vigorously for additional 2 h before being carefully quenched with MeOH. The mixture became slightly warm and clear. The solution was washed with cold water (0° C., 2×150 mL). The washings were extracted back with DCM. The combined org. extracts were again washed with water, dried over MgSO 4, filtered and concentrated. The crude product was purified by CC on silica gel eluting with heptane:EA 1:1 followed by crystallisation from DCM. The purified crystalline product was dried under HIV at 45° C. for 48 h to give the expected product as a white, crystalline powder (1.62 g).
^ Jump up to:ab“Jeraygo EPAR”. European Medicines Agency. 25 April 2024. Archived from the original on 30 April 2024. Retrieved 27 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
Mahfooz K, Najeed S, Tun HN, Khamosh M, Grewal D, Hussain A, et al. (July 2023). “New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension: A Systematic Review and Meta-Analysis”. Current Problems in Cardiology. 48 (7): 101686. doi:10.1016/j.cpcardiol.2023.101686. PMID36893968.