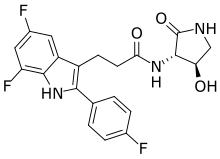
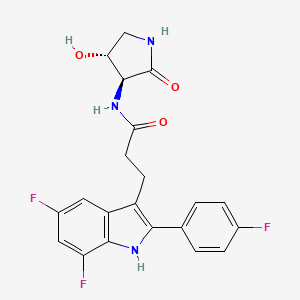
Inaxaplin (VX-147) is a small-molecule apolipoprotein L1 inhibitor developed by Vertex Pharmaceuticals for APOL1-mediated kidney disease. In preliminary studies the drug has shown promise in treating people with kidney disease and multiple gain of function mutations on the APOL1 gene.[1][2]
FSGS is a disease of the podocyte (glomerular visceral epithelial cells) responsible for proteinuria and progressive decline in kidney function. NDKD is a disease characterized by hypertension and progressive decline in kidney function. Human genetics support a causal role for the G1 and G2 APOL1 variants in inducing kidney disease. Individuals with 2 APOL1 risk alleles are at increased risk of developing primary (idiopathic) FSGS, human immunodeficiency virus (HIV)-associated FSGS, and NDKD. Currently, FSGS and NDKD are managed with symptomatic treatment (including blood pressure control using blockers of the renin angiotensin system), and patients with FSGS and heavy proteinuria may be offered high dose steroids. Corticosteroids induce remission in a minority of patients and are associated with numerous side effects. These patients, in particular individuals of recent sub-Saharan African ancestry with 2 APOL1 risk alleles, experience rapid disease progression leading to end-stage renal disease (ESRD). Thus, there is an unmet medical need for treatment for FSGS and NDKD.
Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Method G: Amide Coupling with CDMT. A 2 L 3-neck RB flask with magnetic stirrer, temperature probe and nitrogen inlet was charged with 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic acid S12 (90.5 g, 283.5 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one S2 (39.9 g, 343.6 mmol) in DMF (1.65 L), and stirred for 15 minutes. CDMT (61.1 g, 348 mmol) was added. The mixture was then cooled to −2° C. on an ice bath. N-methylmorpholine was added (131 mL, 1.2 mol) dropwise over 20 minutes and the mixture was heated at 30° C. overnight. The reaction mixture was added into approx. 4.5 L of ice water, and extracted with EtOAc (1.2 L×4). The combined organic layers, were washed with 1.2 L of 1 M HCl (×3) and then water (1.2 L) and brine (1.2 L). The combined organic layers were dried over Na 2SO 4, filtered and concentrated. The mixture was washed through a silica gel plug (1.8 L of silica gel), first eluting with 25% EtOAc in dichloromethane (8 L) to remove impurities, followed by hot EtOAc (8 L), to elute the product. The EtOAc filtrate was concentrated in vacuo. TBME was then added (400 mL), and the mixture allowed to stirred for overnight. Filtration of the resulting solid afforded the product as a white solid. 62 g, 52%) 1H NMR (300 MHz, CD 3OD) δ 7.70-7.58 (m, 2H), 7.29-7.13 (m, 3H), 6.73 (ddd, J=11.1, 9.6, 2.2 Hz, 1H), 4.34 (td, J=7.6, 6.8 Hz, 1H), 4.21 (d, J=7.8 Hz, 1H), 3.56 (dd, J=9.9, 7.6 Hz, 1H), 3.20-3.04 (m, 3H), 2.65-2.53 (m, 2H). LCMS m/z 418.2 [M+H] +. Optical rotation: [α] D20.7=−14.01 (c=1.0, 10 mg in 1 mL of MeOH).
Alternative Procedure for Synthesis of Compound (2)
Step 1. Synthesis of Methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate (C51)
A solution of 5,7-difluoro-2-(4-fluorophenyl)-1H-indole C50 (100 g, 1.0 equiv) in dichloromethane (850 mL, 8.5 vol) was agitated at 22° C. Methyl 3,3-dimethoxypropionate (63 mL, 1.1 equiv) was charged followed by trifluoroacetic acid (96 mL, 3.1 equiv), which was rinsed forward with dichloromethane (25 mL, 0.25 vol). The batch was heated to 38° C. and stirred at that temperature. After 4 h, the batch was cooled to 22° C. and charged with n-heptane (200 mL, 2 vol). The mixture was stirred for no less than 1 h at 22° C. The slurry was filtered, and the reactor and the filter cake were washed with n-heptane (1×2 vol (200 mL) and 1×3 vol (300 mL)). The resulting solid was dried under vacuum with nitrogen bleed at 45° C. to afford the product C51 (127.7 g, 95% yield).
Step 2. Synthesis of Methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate (C52)
To a hydrogenator was charged methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate C51 (100.4 g, 1.0 equiv) followed by Pd(OH) 2/C (0.014 equiv). The vessel was sealed and three vacuum/purge cycles with N2 were performed. 2-MeTHF (2000 mL, 20 vol) was charged using residual vacuum and the resulting mixture was stirred at 22° C. The vessel was sealed and three vacuum/purge cycles with N2 were performed followed by one vacuum purge cycle with hydrogen (H 2). The temperature was adjusted to 22° C., and the vessel pressurized with 20 psi H 2. The mixture was agitated at 22° C. for 4 h. Three vacuum/purge cycles with nitrogen N2 were performed. The batch was filtered through a pad of Hyflo® and the filter cake was rinsed with 2-MeTHF (2×300 mL, 2×3 vol). The combined filtrates were placed under vacuum and distilled at ≤45.0° C. to 2.0 to 3.0 total volumes. The batch temperature was adjusted to 22° C. and the vessel was charge with n-heptane (1000 mL, 10 vol) over at least 1 h. A vacuum was applied and the filtrate distilled at 45.0° C. to 3.5 to 4.5 total volumes. The slurry was cooled to 22° C. and allowed to stir for no less than 1 h. The slurry was filtered and the filter cake was washed with n-heptane (1×1 vol (100 mL) and 1×0.5 vol (50 mL)). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford the product C52 (91.9 g, 91% yield).
Step 3. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic Acid (S12)
A mixture of methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate C52 (80.0 g, 1.0 equiv) and 2-MeTHF (480 mL, 6 vol) was agitated at 22° C. and treated with methanol (72 mL, 0.9 vol). A solution of KOH (27.1 g, 2.0 equiv) in water (107 mL, 1.3 vol) was charged over approximately 20 min. The resulting mixture was heated to an internal temperature of 35° C. and stirred for 3 h. The temperature was adjusted to 22° C. A vacuum was applied and the mixture was distilled at ≤45° C. to 3.0 total volumes. The internal temperature was adjusted to 12° C. The mixture was then charged with water (64 mL, 0.8 vol) and 2-MeTHF (304 mL, 3.8 vol). 6N HCl (75 mL, 0.9 vol) was slowly charged into the mixture with vigorous agitation until the batch attained a pH<3. The internal temperature was adjusted to 22° C., and the biphasic mixture was stirred for no less than 0.5 h. The stirring was stopped and the phases were allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. Water (160 mL, 2 vol) was charged to the reactor at 22° C., and the biphasic mixture stirred for no less than 0.5 h. The stirring was stopped, and the phases allowed separated over no less than 0.5 h. The lower aqueous phase was removed and the batch was filtered through a pad of Hyflo®. The reactor and filter cake were rinsed with 2-MeTHF (160 mL, 2 vol). A vacuum was applied and the combined filtrates distilled at ≤40.0° C. to 2-3 total volumes. The vessel was charged with n-heptane (160 mL, 2 vol), a vacuum was applied and the filtrate distilled at 40.0° C. to 2 total volumes (this step was repeated one additional time). The mixture was then charged with additional n-heptane (144 mL, 1.8 vol). The internal temperature was adjusted to 40° C. and stirred for no less than 2 h. The internal temperature was adjusted to 22° C. over a minimum of 5 h and stirred for no less than 16 hours. The slurry was filtered. The filter cake was washed with n-heptane (3×40 mL, 3×0.5 vol). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford product S12 (72.6 g, 95% yield).
Step 4. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
A mixture of S12 (50.0 g, 1.0 equiv), (3S,4R)-3-amino-4-hydroxypyrrolidin-2-one hydrochloride S2 (25.1 g, 1.05 equiv), and CDMT (30.3 g, 1.1 equiv) in DMF (250 mL, 5 vol) was agitated and cooled to 0° C. The reactor was charged with NMM (60 mL, 3.5 equiv) over no less than 1 h, while maintaining the internal temperature at ≤5° C. The batch was stirred at −5° C. for no less than 1 h. The batch was warmed to 22° C. over at least 1 h and stirred at 22° C. for 16 h. The batch was cooled to 0° C. Water (250 mL, 5 vol) was charged, while keeping the internal temperature <20° C. The mixture was charged with a 90/10 mixture of EtOAc/IPA (1000 mL, 20 vol). 6N HCl (40 mL, 0.8 vol) was then charged, while maintaining an internal temperature <10° C., until a pH˜1-3 was achieved. The internal temperature was adjusted to 22° C. and the biphasic mixture stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. The aqueous layer was back extracted with a 90/10 mixture of EtOAc/IPA (2×250 mL, 2×5 vol) at 22° C. The combined organic phases from extractions were washed with water (5×500 mL, 5×10 vol) at 22° C., by mixing for no less than 0.5 h and settling for no less than 0.5 h for each wash. The batch was polish filtered. A vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes. The mixture was charged with EtOAc (500 mL, 10 vol), vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes (this step was repeated one more time). The mixture was charged with EtOAc (300 mL, 6 vol) and n-heptane (200 mL, 4 vol). The resulting slurry was heated to 50° C. and stirred for no less than 17 h. The mixture was then cooled to 22° C. over 2 h, and stirred for no less than 1 h. The slurry was filtered. The filter cake was washed with 1:1 EtOAc/n-heptane (2×150 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (52.6 g, 80% yield).
Re-Crystallization of Compound 2
Compound 2 (37.6 g, 1.0 equiv) was charged to a reactor followed by a 3:1 mixture of IPA/water (240 mL, 6.4 vol). The slurry was heated to an internal temperature of 75° C. The batch was cooled to an internal temperature of 55° C. and stirred at that temperature for at least 0.5 h. The batch was seeded with 0.5 wt % of a previously generated batch of Compound 2, as a suspension in a mixture of 3:1 IPA/water (4 mL, 0.1 vol). The mixture was stirred at 55° C. for no less than 1.5 h. Water (218 mL, 5.8 vol) was added over minimum period of 5 h while maintaining the temperature at 55° C. The slurry was cooled to 22° C. over no less than 5 h and stirred for no less than 2 h. The slurry was filtered. The filter cake was washed with 2:3 IPA/water (2×114 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (34.5 g, 92% yield).
Form A of Compound 2
12.3 kg of Compound 2 was charged to the reactor follow by a 3:1 mixture of 2-propanol/water. Agitation was initiated and the mixture was heated to 75° C. to achieve complete dissolution. The mixture was cooled to 55° C. over 1 hour and agitated at that temperature for 30 minutes. Agitation was continued for 1.5 hours. Water (5.8 vol) was charged over 5 h at 55° C., after which the mixture was cooled to 22° C. over 6 hours. The mixture was agitated at 22° C. for 2 hours then filtered under vacuum. The resulting wet cake was washed with a 3:1 mixture of 2-propanol/water (2.74 vol×2) and pulled dry under vacuum. The wet cake was further dried under vacuum with nitrogen bleed at 45° C. to yield 11.2 kg of Form A.
Hydrate Form A of Compound 2
200 mg of Compound 2 was charged with 10 mL of water. The slurry was cooled to 5° C. and allowed to stir. Hydrate A was observed after 3 days of stirring.