
![Chemical structure of gadopiclenol [gadolinium chelate of 2,2′,2″-(3,6,9-triaza-1(2,6)-pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid)]. The PCTA parent structure is shown in red. Two water molecules are included to show the coordination in solution.](http://www.researchgate.net/profile/Jean-Marc-Idee/publication/334838366/figure/fig1/AS:797490152476678@1567147877999/Chemical-structure-of-gadopiclenol-gadolinium-chelate-of.jpg)
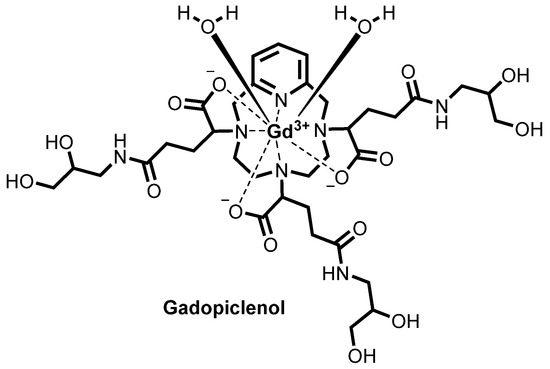
Gadopiclenol
ガドピクレノール;
| Formula | C35H54N7O15. Gd |
|---|---|
| CAS | 933983-75-6 |
| Mol weight | 970.0912 |
FDA APPROVED 2022/9/21, Elucirem
Diagnostic agent (MR imaging), WHO 10744, P 03277, UNII: S276568KOY
EluciremTM; G03277; P03277; VUEWAY
(alpha3,alpha6,alpha9-Tris(3-((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca-1(15),11,13-triene-3,6,9-triacetato(3-)-kappaN3,kappaN6,kappaN9,kappaN15,kappaO3,kappaO6,kappaO9)gadolinium
- OriginatorGuerbet
- ClassDiagnostic agents; Gadolinium-containing contrast agents; Macrocyclic compounds; Propylamines; Pyridines
- Mechanism of ActionMagnetic resonance imaging enhancers
- RegisteredCNS disorders
- Phase IIIUnspecified
- Phase IILiver cancer
- 21 Sep 2022Registered for CNS disorders (Diagnosis) in USA (IV)
- 13 Jun 2022Guerbet plans to launch Gadopiclenol in Europe
- 13 Jun 2022The European Medicines Agency (EMA) accepts brand name EluciremTM for Gadopiclenol
PATENT
https://patents.google.com/patent/WO2020030618A1/en
MRI contrast agents used in daily diagnostic practice typically include gadolinium complex compounds characterized by high stability constants that guarantee against the in vivo release of the free metal ion (that is known to be extremely toxic for living organisms).
Another key parameter in the definition of the tolerability of a gadolinium-based contrast agent is the kinetic inertness (or kinetic stability) of Gd(III)-complex, that is estimated through the half-life (ti/2) of the dissociation (i.e. decomplexation) of the complex.
A high inertness becomes crucial in particular for those complex compounds having lower thermodynamic stability and/or longer retention time before excretion, in order to avoid or minimize possible decomplexation or transmetallation reactions.
EP1931673 (Guerbet) discloses PCTA derivatives of formula

and a synthetic route for their preparation.
EP 2988756 (same Applicant) discloses a pharmaceutical composition comprising the above derivatives together with a calcium complex of 1,4,7, 10-tetraazacyclododecane- 1,4,7, 10-tetraacetic acid. According to the EP 2988756, the calcium complex compensates the weak thermodynamic stability observed for PCTA-based gadolinium complexes, by forming, through transmetallation, a strong complex with free lanthanide ion, thereby increasing the tolerability of the contrast agent.
Both EP1931673 and EP 2988756 further refer to enantiomers or diastereoisomers of the claimed compounds, or mixture thereof, preferentially chosen from the RRS, RSR, and RSS diastereoisomers. Both the above patents disclose, among the specific derivatives, (a3, a6, a9)-tris(3- ((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca- l(15),l l,13-triene-3,6,9-triacetato(3-)-(KN3,KN6,KN9,KN15,K03,K06,K09)gadolinium, more recently identified as gadolinium chelate of 2,2′,2″-(3,6,9-triaza-l(2,6)- pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid), (CAS registry number: 933983-75-6), having the following formula

otherwise identified as P03277 or Gadopiclenol.
For Gadopiclenol, EP1931673 reports a relaxivity of 11 mM _1s _1Gd 1 (in water, at 0.5 T, 37°C) while EP 2988756 reports a thermodynamic equilibrium constant of 10 14 9 (log Kterm
= 14.9).
Furthermore, for this same compound a relaxivity value of 12.8 mM _1s 1 in human serum (37°C, 1.41 T), stability (log Kterm) of 18.7, and dissociation half-life of about 20 days (at pH 1.2; 37°C) have been reported by the proprietor (Investigative Radiology 2019, Vol 54, (8), 475-484).
The precursor for the preparation of the PCTA derivatives disclosed by EP1931673 (including Gadopiclenol) is the Gd complex of the 3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),l l,13-triene-tri(a-glutaric acid) having the following formula

Gd(PCTA-tris-glutaric acid)
herein identified as “Gd(PCTA-tris-glutaric acid)”. In particular, Gadopiclenol is obtained by amidation of the above compound with isoserinol.
As observed by the Applicant, Gd(PCTA-tris-qlutaric acid) has three stereocenters on the glutaric moieties (identified with an asterisk (*) in the above structure) that lead to a 23 = 8 possible stereoisomers. More particularly, the above structure can generate four pairs of enantiomers, schematized in the following Table 1
Table 1

Isomer RRR is the mirror image of isomer SSS and that is the reason why they are called enantiomers (or enantiomer pairs). As known, enantiomers display the same physicochemical properties and are distinguishable only using chiral methodologies, such as chiral chromatography or polarized light.
On the other hand, isomer RRR is neither equal to nor is it the mirror image of any of the other above six isomers; these other isomers are thus identified as diastereoisomers of the RRR (or SSS) isomer. Diastereoisomers may display different physicochemical properties, (e.g., melting point, water solubility, relaxivity, etc.).
Concerning Gadopiclenol, its chemical structure contains a total of six stereocenters, three on the glutaric moieties of the precursor as above discussed and one in each of the three isoserinol moieties attached thereto, identified in the following structure with an asterisk (*) and with an empty circle (°), respectively:

This leads to a total theoretical number of 26 = 64 stereoisomers for this compound. However, neither EP1931673 nor EP 2988756 describe the exact composition of the isomeric mixture obtained by following the reported synthetic route, nor does any of them provide any teaching for the separation and characterization of any of these isomers, or disclose any stereospecific synthesis of Gadopiclenol. Summary of the invention
The applicant has now found that specific isomers of the above precursor Gd(PCTA- tris-glutaric acid) and of its derivatives (in particular Gadopiclenol) possess improved physico-chemical properties, among other in terms of relaxivity and kinetic inertness.
An embodiment of the invention relates to a compound selected from the group consisting of:
the enantiomer [(aR,a’R,a”R)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium (RRR enantiomer) having the formula (la):

the enantiomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),ll,13-triene-3,6,9-triacetato(3-)KN3,KN6,KN9,KN15,K03,K06,K09]- gadolinium (SSS enantiomer) having the formula (lb):

the mixtures of such RRR and SSS enantiomers, and a pharmaceutically acceptable salt thereof.
Another embodiment of the invention relates to an isomeric mixture of Gd(PCTA-tris- glutaric acid) comprising at least 50% of the RRR isomer [(aR,a’R,a”R)-a,a’,a”-tris(2- carboxyethyl)-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9- triacetato(3-)-KN3,KN6,KN9,KN15,K03,K06,K09]-gadolinium, of formula (la), or of the SSS isomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium of formula (lb), or of a mixture thereof, or a pharmaceutically acceptable salt thereof. Another aspect of the invention relates to the amides obtained by conjugation of one of the above compounds or isomeric mixture with an amino group, e.g. preferably, serinol or isoserinol.
An embodiment of the invention relates to an amide derivative of formula (II A)
F( N RI R2)3 (II A)
in which :
F is:
a RRR enantiomer residue of formula Ilia

a SSS enantiomer residue of formula Illb

or a mixture of such RRR and SSS enantiomer residues;
and each of the three -NRIR2 group is bound to an open bond of a respective carboxyl moiety of F, identified with a full circle (·) in the above structures;
Ri is H or a Ci-Ce alkyl, optionally substituted by 1-4 hydroxyl groups;
R2 is a Ci-Ce alkyl optionally substituted by 1-4 hydroxyl groups, and preferably a C1-C3 alkyl substituted by one or two hydroxyl groups.
Another embodiment of the invention relates to an isomeric mixture of an amide derivative of Gd(PCTA-tris-glutaric acid) having the formula (II B)
F'( N RI R2)3 (II B)
in which :
F’ is an isomeric mixture of Gd(PCTA-tris-glutaric acid) residue of formula (III)

said isomeric mixture of the Gd(PCTA-tris-glutaric acid) residue comprising at least 50 % of an enantiomer residue of the above formula (Ilia), of the enantiomer residue of the above formula (Illb), or of a mixture thereof; and each of the -NR1R2 groups is bound to an open bond of a respective carboxyl moiety of F’, identified with a full circle (·) in the above structure, and is as above defined for the compounds of formula (II A).
EXPERIMENTAL PART
HPLC characterization of the obtained compounds.
General procedures
Procedure 1: HPLC Characterization of Gd(PCTA-tris-glutaric acid) (isomeric mixture and individual/enriched isomers).
The HPLC characterization of the Gd(PCTA-tris-glutaric acid) obtained as isomeric mixture from Example 1 was performed with Agilent 1260 Infinity II system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector ( Agilent 1260 Infinity II system)
Stationary phase: Phenomenex Gemini® 5pm C18 lloA
Mobile phase: H2O/HCOOH 0.1% : Methanol
Elution : Gradient Time (min) H2O/HCOOH 0.1% Methanol
0 95 5
5 95 5
30 50 50
35 50 50
40 95 5
Flow 0.6 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
Injection volume 50 pL
Sample Cone. 0.2 mM Gd(PCTA-tris-glutaric acid) complex
Stop time 40 min
Retention time GdL = 18-21 min.
Obtained HPLC chromatogram is shown in Figure 1
The HPLC chromatogram of the enriched enantiomers pair C is shown in Figure 2.
Procedure 2: HPLC Characterization of Gadopiclenol (isomeric mixture) and compounds obtained by coupling of enantiomers pair C with R, S, or racemic isoserinol.
The HPLC characterization of Gadopiclenol either as isomeric mixture obtained from Example 2, or as the compound obtained by conjugation of enantiomers pair C of the Gd(PCTA-tris-glutaric acid) with R, S, or racemic isoserinol was performed with Thermo Finnigan LCQ DECA XPPIus system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA and MS detector (LCQ Deca XP-Plus – Thermo Finnigan )
Stationary phase: Phenomenex Gemini 5u C18 110A
Mobile phase: H2O/TFA 0.1% : Acetonitrile/0.1%TFA
Elution : Gradient Time (min) H2O/TFA 0.1% Acetonitrile/0.1%TFA
0 100 0
5 100 0
22 90 10
26 90 10
Flow 0.5 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
MS positive mode – Mass range 100-2000
Injection volume 50 pL
Sample cone. 0.2 mM Gd complex
Stop time 26 min
Retention time GdL = 20-22min.
Obtained HPLC chromatograms are shown in Figure 6.
Procedure 3: Chiral HPLC method for the separation of enantiomers of the compound C
A specific chiral HPLC method was set up in order to separate the RRR and SSS enantiomers of the enantiomers pair C (compound VI), prepared as described in Example 3. The separation and characterization of the enantiomers were performed with Agilent 1200 system or Waters Alliance 2695 system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC System HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector
Stationary phase SUPELCO Astec CHIROBIOTIC 5 pm 4.6x250mm
Mobile phase H2O/HCOOH 0.025% : Acetonitrile
Elution : isocratic 2% Acetonitrile for 30 minutes
Flow 1 mL/min
Column Temperature 40°C
Detection 210-270 nm. Obtained HPLC chromatogram is shown in Figure 5a) compared to the chromatograms of the pure RRR enantiomer (compound XII of Example 5, Tr. 7.5 min.) and the pure SSS enantiomer (Compound XVII of Example 6, Tr. 8.0 min), shown in figure 5b) and 5c), respectively.
Example 1: Synthesis of Gd(PCTA-tris-glutaric acid) (isomeric mixture)
Gd(PCTA-tris-glutaric acid) as an indiscriminate mixture of stereoisomers has been prepared by using the procedure reported in above mentioned prior-art, according to the following synthetic Scheme 1 :
Scheme 1

a) Preparation of Compound II
Racemic glutamic acid (33.0 g, 0.224 mol) and sodium bromide (79.7 g, 0.782 mol) were suspended in 2M HBr (225 ml_). The suspension was cooled to -5°C and NaN02 (28.0 g, 0.403 mol) was slowly added in small portions over 2.5 hours, maintaining the inner temperature lower than 0 °C. The yellow mixture was stirred for additional 20 minutes at a temperature of -5°C; then concentrated sulfuric acid (29 ml.) was dropped in the mixture. The obtained dark brown mixture was warmed to RT and then extracted with diethyl ether (4×150 ml_). The combined organic phases were washed with brine, dried over Na2S04 and concentrated to a brown oil (21.2 g), used in the following step without further purification. The oil was dissolved in ethanol (240 ml_), the resulting solution was cooled in ice and thionyl chloride (14.5 ml_, 0.199 mol) was slowly added. The slightly yellow solution was stirred at RT for 2 days. Then the solvent was removed in vacuum and the crude oil was dissolved in dichloromethane (200 ml.) and washed with 5% aq. NaHCC>3 (4×50 ml_), water (1×50 ml.) and brine (1×50 ml_). The organic phase was concentrated and purified on silica eluting with petroleum ether-ethyl acetate 3: 1, obtaining 19.5 g of pure product. (Yield 33%).
b) Preparation of Compound IV
A solution of Compound II (17.2 g, 0.0645 mol) in acetonitrile (40 ml.) was added to a suspension of 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene (pyclen) Compound (III) (3.80 g, 0.018 mol) and K2CO3 (11.2 g, 0.0808 mol) in acetonitrile (150 ml_). The yellow suspension was heated at 65 °C for 24 h, then the salts were filtered out and the organic solution was concentrated. The orange oil was dissolved in dichloromethane and the product was extracted with 1M HCI (4 x 50 ml_). The aqueous phases were combined, cooled in ice and brought to pH 7-8 with 30% aq. NaOH. The product was then extracted with dichloromethane (4 x 50 ml.) and concentrated to give a brown oil (10.1 g, yield 73%). The compound was used in the following step without further purification.
c) Preparation of compound V
Compound IV (9.99 g, 0.013 mol) was dissolved in Ethanol (40 ml.) and 5M NaOH (40 ml_). The brown solution was heated at 80 °C for 23 h. Ethanol was concentrated; the solution was cooled in ice and brought to pH 2 with cone HCI. The ligand was purified on resin Amberlite XAD 1600, eluting with water-acetonitrile mixture, obtaining after freeze- drying 5.7 g as white solid (yield 73%). The product was characterized in HPLC by several peaks.
d) Preparation of compound VI
Compound V (5.25 g, 0.0088 mol) was dissolved in deionized water (100 ml.) and the solution was brought to pH 7 with 2M NaOH (20 ml_). A GdCh solution (0.0087 mol) was slowly added at RT, adjusting the pH at 7 with 2M NaOH and checking the complexation with xylenol orange. Once the complexation was completed, the solution was concentrated and purified on resin Amberlite XAD 1600 eluting with water-acetonitrile gradient, in order to remove salts and impurities. After freeze-drying the pure compound was obtained as white solid (6.79 g, yield 94%). The product was characterized in HPLC; the obtained HPLC chromatogram, characterized by several peaks, is shown in Figure 1 A compound totally equivalent to compound VI, consisting of an isomeric mixture with a HPLC chromatogram substantially superimposable to that of Figure 1 is obtained even by using (S)-methyl a-bromoglutarate obtained starting from L-glutamic acid.
Example 2: Synthesis of Gadopiclenol (isomeric mixture)
Gadopiclenol as an indiscriminate mixture of stereoisomers has been prepared as disclosed in EP11931673 B1 by coupling the isomeric mixture of Gd(PCTA-tris-glutaric acid) obtained from Example 1 with racemic isoserinol according to the following synthetic Scheme 2:
Scheme 2

Preparation of compound VII
Compound VI (0.90 g, 0.0011 mol) obtained from Example 1 was added to a solution of racemic isoserinol (0.40 g, 0.0044 mol) in water adjusted to pH 6 with cone. HCI. Then N- ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI-HCI) (1.0 g, 0.0055 mol) and hydroxybenzotriazole (HOBT) (0.12 g, 0.00088 mol) were added and the resulting solution was stirred at pH 6 and RT for 24 h. The product was then purified on preparative HPLC on silica C18, eluting with water/acetonitrile gradient. Fractions containing the pure compound were concentrated and freeze-dried, obtaining a white solid (0.83 g, yield 78%). The product was characterized in HPLC; the obtained HPLC chromatogram is shown in Figure 4a.
Example 3: Isolation of the enantiomers pair related to the peak C.
Compound VI obtained as described in Example 1 (step d) (1.0 g, 0.0013 mol) was dissolved in water (4 ml.) and the solution was acidified to pH 2-3 with cone. HCI. The obtained solution was loaded into a pre-packed column of silica C18 (Biotage® SNAP ULTRA C18 120 g, HP-sphere C18 25 pm) and purified with an automated flash chromatography system eluting with deionized water (4 CV) and then a very slow gradient of acetonitrile. Fractions enriched of the enantiomers pair related to the peak C were combined, concentrated and freeze-dried obtaining a white solid (200 mg).
The HPLC chromatogram of the obtained enriched enantiomers pair C is shown in Figure 2.
Corresponding MS spectrum (Gd(H4L)+:752.14 m/z) is provided in Figure 3
Example 4: Coupling of the enantiomers pair C with isoserinol.
a) Coupling of the enantiomers pair C with R-isoserinol.
Enriched enantiomers pair C collected e.g. as in Example 3 (34 mg, titer 90%, 0.040 mmol) was dissolved in deionized water (5 ml_), and R-isoserinol (16 mg, 0.17 mmol) was added adjusting the pH at 6 with HCI 1M. Then, EDCI-HCI (39 mg, 0.20 mmol) and HOBT (3 mg, 0.02 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (21 mg, yield 54%).
The HPLC chromatogram of the obtained product is shown in Figure 6b.
b) Coupling of the enantiomers pair C with S-isoserinol
Enriched enantiomers pair C collected e.g. as in Example 3 (55 mg, titer 90%, 0.066 mmol) was dissolved in deionized water (5 mL), and S-isoserinol (34 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (64 mg, 0.33 mmol) and HOBT (4.5 mg, 0.033 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (52 mg, yield 81%).
HPLC chromatogram of the obtained product is shown in Figure 6c.
c) Coupling of the enantiomers pair C with racemic isoserinol.
The enriched enantiomers pair C collected e.g. as in Example 3 (54 mg, titer 90%, 0.065 mmol) was dissolved in deionized water (5 mL), and racemic isoserinol (27 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (62 mg, 0.32 mmol) and HOBT (4.3 mg, 0.032 mmol) were added and the solution was stirred at RT at pH 6 for 24 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (60 mg, yield 95%).
HPLC chromatogram of the obtained product is shown in Figure 6d. Example 5: Stereoselective synthesis of the RRR Gd(PCTA-tris-glutaric acid) (compound XII).
RRR enriched Gd(PCTA-tris-glutaric acid) acid has been prepared by following the synthetic Scheme 3 below
Scheme 3

comprising :
a) Preparation of Compound VIII
The preparation was carried out as reported in Tetrahedron 2009, 65, 4671-4680.
In particular: 37% aq. HCI (50 pL) was added to a solution of (S)-(+)-5- oxotetrahydrofuran-2-carboxylic acid (2.48 g, 0.019 mol) (commercially available) in anhydrous methanol (20 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHCC>3 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (2.97 g, yield 89%).
b) Preparation of Compounds IX and X
Compound VIII (445 mg, 2.52 mmol) obtained at step a) was dissolved in anhydrous dichloromethane (6 ml.) and triethylamine (0.87 ml_, 6.31 mmol) was added. The solution was cooled at -40°C and then (triflic) trifluoromethansulfonic anhydride (0.49 ml_,2.91 mmol) was slowly added. The dark solution was stirred at -40°C for 1 h, then a solution of Compound III (104 mg, 0.506 mmol) in anhydrous dichloromethane (3 ml.) and triethylamine (1 ml_, 7.56 mmol) were added and the solution was slowly brought to RT and stirred at RT overnight. The organic solution was then washed with 2M HCI (4x 10 ml_), the aqueous phase was extracted again with dichloromethane (3 x 10 ml_). The organic phases were combined and concentrated in vacuum, obtaining 400 mg of a brown oil that was used in the following step with no further purification.
c) Preparation of Compound XI
Compound X (400 mg, 0.59 mmol) was dissolved in methanol (2.5 ml.) and 5M NaOH (2.5 ml_). The brown solution was heated at 80°C for 22 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 1 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (64 mg, yield 18 %). The HPLC showed a major peak.
d) Compound XII
Compound XI (32 mg, 0.054 mmol) was dissolved in deionized water (4 mL) and the pH was adjusted to 7 with 1M NaOH. GdCl3-6H20 (20 mg, 0.054 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The clear solution was stirred at RT overnight and the end of the complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired RRR isomer as major peak: about 80% in area %. The mixture was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (36 mg, yield 90%).
By reaction of the collected compound with isoserinol e.g. by using the procedure of the Example 2, the corresponding RRR amide derivative can then be obtained.
Example 6: stereoselective synthesis of the SSS Gd(PCTA-tris-glutaric acid) (compound XVII).
SSS enriched Gd(PCTA-tris-glutaric acid) acid has been similarly prepared by following the synthetic Scheme 4 below Scheme 4

comprising :
a) Preparation of Compound XIII
37% aq. HCI (100 pl_) was added to a solution of (R)-(-)-5-oxotetrahydrofuran-2- carboxylic acid (5.0 g, 0.038 mol) (commercially available) in anhydrous methanol (45 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHC03 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (6.7 g, yield 99%).
b) Preparation of Compounds XIV and XV
Compound XIII (470 mg, 2.67 mmol) was dissolved in anhydrous dichloromethane (6 ml.) and trimethylamine (0.93 ml_, 6.67 mmol) was added. The solution was cooled down at -40°C and then trifluoromethanesulfonic anhydride (0.50 ml_, 3.07 mmol) was slowly dropped. The dark solution was stirred at -40°C for 1 h, then Compound III (140 mg, 0.679 mmol) and trimethylamine (0.93 ml_, 6.67 mmol) were added and the solution was slowly brought to RT overnight. The organic solution was then washed with water (3 x 5 ml.) and 2M HCI (4 x 5 ml_). The aqueous phase was extracted again with dichloromethane (3 x 10 ml_). the organic phases were combined and concentrated in vacuum, obtaining 350 mg of a brown oil that was used in the following step with no further purification. c) Preparation of Compound XVI
Compound XV (350 mg, 0.514 mmol) was dissolved in methanol (4.5 ml.) and 5M NaOH (4.5 ml_). The obtained brown solution was heated at 80°C for 16 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (52 mg, yield 17%). The HPLC showed a major peak.
d) Preparation of Compound XVII
Compound XVI (34 mg, 0.057 mmol) was dissolved in deionized water (5 mL) and the pH was adjusted to 7 with 1 M HCI. GdCl3-6H20 (20 mg, 0.0538 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The solution was stirred at RT overnight and the end of complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired SSS isomer as major peak: about 85% in area %. The solution was brought to pH 2.5 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product SSS were combined, concentrated and freeze-dried (39 mg, yield 87%).
Example 7: Kinetic studies of the dissociation reactions of Gd(PCTA-tris- glutaric acid) (isomeric mixture) in 1.0 M HCI solution (25°C)
The kinetic inertness of a Gd(III)-complex is characterized either by the rate of dissociation measured in 0.1-1.0 M HCI or by the rate of the transmetallation reaction, occurring in solutions with Zn(II) and Cu(II) or Eu(III) ions. However, the dissociation of lanthanide(III)-complexes formed with macrocyclic ligands is very slow and generally proceeds through a proton-assisted pathway without the involvement of endogenous metal ions like Zn2+ and Cu2+.
We characterized the kinetic inertness of the complex Gd(PCTA-tris-glutaric acid) by the rates of the dissociation reactions taking place in 1.0 M HCI solution. The complex (isomeric mixture from Example 1) (0.3 mg) was dissolved in 2.0 mL of 1.0 M HCI solution and the evolution of the solution kept at 25 °C was followed over time by HPLC. The HPLC measurements were performed with an Agilent 1260 Infinity II system by use of the analytical Procedure 1.
The presence of a large excess of H+ ([HCI] = 1.0 M), guarantees the pseudo-first order kinetic conditions.
GdL + yH÷ ^ Gd3+ + HyL y=7 and 8 (Eg. 1) where L is the protonated PCTA-tri-glutaric acid, free ligand, and y is the number of protons attached to the ligand.
The HPLC chromatogram of Gd(PCTA-tris-glutaric acid) is characterized by the presence of four signals (A, B, C and D) having the same m/z ratio (Gd(H4L)+ :752.14 m/z) in the MS spectrum. Each of these peaks is reasonably ascribable to one of the 4 pairs of enantiomers generated by the three stereocenters on the three glutaric arms of the molecule, formerly identified in Table 1. The HPLC chromatogram of this complex in the presence of 1.0 M HCI changes over time: in particular, the areas of peaks A, B, C, and D decrease, although not in the same way for the different peaks, while new signals corresponding to non-complexed diastereoisomers are formed and grow over time. Differences in the decrease of the integral areas of the peaks can be interpreted by a different dissociation rate of the enantiomer pairs associated to the different peaks.
In the presence of [H + ] excess the dissociation reaction of enantiomer pairs of Gd(PCTA-tris-glutaric acid) can be treated as a pseudo-first-order process, and the rate of the reactions can be expressed with the following Eq. 2, where kA, kB, kc and kD are the pseudo-first-order rate constants that are calculated by fitting the area-time data pair, and [A]t, [B]t, [C]t and [D]t are the total concentration of A, B, C and D compounds at time t.

The decrease of the area values of signals of A, B, C, and D has been assessed and plotted over time. Area values of A, B, C and D signals as a function of time are shown in Figure 7.
Area value at time t can be expressed by the following equation:
A. = A + (A0 – A )e kxt
(Eg. 3)
where At, A0 and Ae are the area values at time t, at the beginning and at the end of the reactions, respectively, kx pseudo-first-order rate constants (/fX=/fA, kB, kc and kD) characterizing the dissociation rate of the different enantiomer pairs of Gd(PCTA-tris-glutaric acid) complex were calculated by fitting the area – time data pairs of Figure 7 to the above equation 3. kx rate constants and half-lives (ti/2= In2/ x) are thus obtained, as well as the average the half-life value for the isomeric mixture of Gd(PCTA-tris-glutaric acid), calculated by considering the percentage composition of the mixture. Obtained values are summarized in the following Table 2, and compared with corresponding values referred in the literature for some reference contrast agents. (Gd-DOTA or DOTAREM ). Table 2. Rate constants ( kx ) and half-lives (ti/2= In2/ x) characterizing the acid catalyzed dissociation of the different stereoisomers of Gd(PCTA-tris-glutaric acid), Dotarem® and Eu(PCTA) in 1.0 M HCI (pH 0) ( 25°C)
). Table 2. Rate constants ( kx ) and half-lives (ti/2= In2/ x) characterizing the acid catalyzed dissociation of the different stereoisomers of Gd(PCTA-tris-glutaric acid), Dotarem® and Eu(PCTA) in 1.0 M HCI (pH 0) ( 25°C)
A B C D
Ms 1) (4.5±0.1) x105 (1.1±0.1)x104 (1.6±0.1)x10-6 (1.2±0.1)x10-5 fi/2 (hour) 4.28 ± 0.03 1.76 ± 0.02 120 ± 3 15.8 ± 0.5
fi/2 (hour)

average
Dotarem a
k, (S‘1) 8.0×10-6
fi/2 (hour) 23 hour
Eu(PCTA) b
*1 (s·1) 5.08X10·4
fi/2 (hour) 0.38 hour
a) Inorg. Chem. 1992, 31 ,1095-1099.
b) Tircso, G. et al. Inorg Chem 2006, 45 (23), 9269-80.
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
A gadolinium-based paramagnetic contrast agent, with potential imaging enhancing activity upon magnetic resonance imaging (MRI). Upon administration of gadopiclenol and placement in a magnetic field, this agent produces a large magnetic moment and creates a large local magnetic field, which can enhance the relaxation rate of nearby protons. This change in proton relaxation dynamics, increases the MRI signal intensity of tissues in which this agent has accumulated; therefore, contrast and visualization of those tissues is enhanced compared to unenhanced MRI.
FDA Approves New MRI Contrast Agent Gadopiclenol
September 22, 2022
https://www.diagnosticimaging.com/view/fda-approves-new-mri-contrast-agent-gadopiclenol
Requiring only half of the gadolinium dose of current non-specific gadolinium-based contrast agents (GBCAs), gadopiclenol can be utilized with magnetic resonance imaging (MRI) to help detect lesions with abnormal vascularity in the central nervous system and other areas of the body.
Gadopiclenol, a new magnetic resonance imaging (MRI) contrast agent that offers high relaxivity and reduced dosing of gadolinium, has been approved by the Food and Drug Administration (FDA).1
Approved for use with MRI in adults and pediatric patients two years of age or older, gadopiclenol is a macrocyclic gadolinium-based contrast agent that aids in the diagnosis of lesions with abnormal vascularity in the brain, spine, abdomen, and other areas of the body.
Recently published research demonstrated that gadopiclenol provides contrast enhancement and diagnostic efficacy at half of the gadolinium dosing of other gadolinium-based contrast agents (GBCAs) such as gadobutrol and gadobenate dimeglumine.2
Co-developed by Bracco Diagnostics and Guerbet, gadopiclenol will be manufactured and marketed as Vueway (Bracco Diagnostics) and Elucirem (Guerbet).1,3
Alberto Spinazzi, M.D., the chief medical and regulatory officer at Bracco Diagnostics, said gadopiclenol is “a first of its kind MRI agent that delivers the highest relaxivity and highest kinetic stability of all GBCAs on the market today.”
Reference
1. Bracco Diagnostics. Bracco announces FDA approval of gadopiclenol injection, a new macrocyclic high-relaxivity gadolinium-based contrast agent which will be commercialized as VUEWAY (gadopiclenol) injection and VUEWAY (gadopiclenol) phamarcy bulk package by Bracco. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/bracco-announces-fda-approval-of-gadopiclenol-injection-a-new-macrocyclic-high-relaxivity-gadolinium-based-contrast-agent-which-will-be-commercialized-as-vueway-gadopiclenol-injection-and-vueway-gadopiclenol-pharmacy-bulk-p-301630124.html . Published September 21, 2022. Accessed September 21, 2022.
2. Bendszus M, Roberts D, Kolumban B, et al. Dose finding study of gadopiclenol, a new macrocyclic contrast agent, in MRI of central nervous system. Invest Radiol. 2020;55(3):129-137.
3. Guerbet. Guerbet announces U.S. Food and Drug Administration (FDA) approval of Elucirem (gadopiclenol) injection for use in contrast-enhanced MRI. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/guerbet-announces-us-food-and-drug-administration-fda-approval-of-elucirem-gadopiclenol-injection-for-use-in-contrast-enhanced-mri-301630085.html . Published September 21, 2022. Accessed September 21, 2022.
////Gadopiclenol, FDA 2022, APPROVALS 2022, ガドピクレノール, WHO 10744, P 03277, EluciremTM, G03277; P03277, VUEWAY, Guerbet
