
Crenolanib
- Molecular FormulaC26H29N5O2
- Average mass443.541 Da
1-(2-{5-[(3-Methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl}-8-quinolinyl)-4-piperidinamine
1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine
1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-4-piperidinamine
4-Piperidinamine, 1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-
670220-88-9[RN]
9459
UNII-LQF7I567TQ
креноланиб
كرينولانيب
克拉尼布
CP-868,596-26 or AR-868,596-26
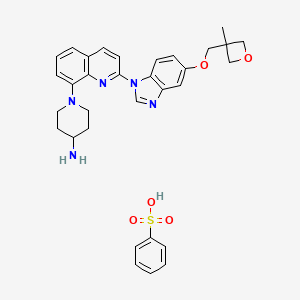
Crenolanib besylate
CAS#: 670220-93-6 (besylate)
Chemical Formula: C32H35N5O5S
Molecular Weight: 601.72
Crenolanib besylate (CP-868,596-26 or AR-868,596-26, 4-piperidinamine, 1-[2-[5-[(3-Methyl-3-oxetanyl) methoxy]-1H-benzimidazol-1-yl]- 8-quinolinyl]-, monobenzenesulfonate) is an investigational inhibitor being developed by AROG Pharmaceuticals, LLC. The compound is currently being evaluated for safety and efficacy in clinical trials for various types of cancer, including acute myeloid leukemia (AML),[1][2] gastrointestinal stromal tumor (GIST),[3] and glioma.[4] Crenolanib is an orally bioavailable benzamidazole that selectively and potently inhibits signaling of wild-type and mutant isoforms of class III receptor tyrosine kinases (RTK) FLT3 (FMS-like Tyrosine Kinase 3), PDGFR α (Platelet-Derived Growth Factor Receptor), and PDGFR β. Unlike most RTK inhibitors, crenolanib is a type I mutant-specific inhibitor that preferentially binds to phosphorylated active kinases with the ‘DFG in’ conformation motif.[5]
CN 109678849

PATENT
WO/2022/060421CRENOLANIB FOR TREATING TRK KINASE ASSOCIATED PROLIFERATIVE DISORDERS
PATENT
WO/2022/060422CRENOLANIB FOR TREATING PAIN
PAPER
https://www.nature.com/articles/s41598-018-21839-3
PAPER
Chembiochem : a European journal of chemical biology (2019), 20(14), 1783-1788.
PATENT
CN 109678849
PATENT
WO 2018118598
https://patents.google.com/patent/WO2018118598A1/en
PAT
US 20170121321
PAT
CN 107382984
https://patents.google.com/patent/CN107382984A/en
Embodiment is as follows:
The synthesis of the chloro- 8- trifluoromethanesulfonic acids base quinoline (Ι) of 2-
Compound 2- chloro-8-hydroxyquinolines 50g, DMF150ml, trifluoromethanesulfchloride chloride 53g, triethylamine 25g are added to 250ml In three-necked bottle, stir.Temperature control reacts 20~30h at 25~30 DEG C.After reaction completely, the solid of precipitation is filtered, filter cake is used Wash washing, 40 DEG C of forced air dryings, the chloro- 8- trifluoromethanesulfonic acids base benzimidazoles of gained off-white powder 2- in n-hexane 20ml × 3 83.39g yield 95.78%.
The synthesis of (base of piperidines -4) the quinoline t-butyl carbamates of 2- chloro- 8 (II)
BINAP 0.2g, toluene 70ml are added into 250ml three-necked bottles, temperature control stirs 1h at 20~25 DEG C.Added again into bottle The chloro- 8- trifluoromethanesulfonic acids base quinoline 10g of 2-, piperidin-4-yl t-butyl carbamate 6.41g, potassium carbonate 7.8g, stir lower by instead Answer liquid to be heated to 80 DEG C~100 DEG C, keep 20~30h of this thermotonus.TLC is detected, and whether reaction is complete.Reaction is complete, stops Only heat.20~30 DEG C are cooled to, dichloroethanes 50ml is added, adds diatomite to filter out the solid in reaction solution, filter cake second Acetoacetic ester 150ml is washed., 20~25 DEG C of stirring 8h.The solid separated out in solution is filtered out, filtrate is molten with 5% disodium hydrogen phosphate Liquid 2x50ml is washed.Organic phase is concentrated to dryness again, adds acetonitrile 50ml, 20~25 DEG C of 10~20h of stirring and crystallizing.Filtering analysis The solid gone out, 40 DEG C of forced air dryings obtain the tertiary fourth of yellow solid 10.69g, 2- chloro- 8 (piperidin-4-yl) benzimidazole carbamic acid Ester, yield 92.3%.
The synthesis of 5- (3- methy oxetane -3- methoxyl groups) benzimidazole (III)
Compound 3- methyl -3- oxetane methanols 30.77g, THF140ml, metallic sodium 6.95g are added to the necks of 250ml tri- In bottle, 66 DEG C of backflow 4h are heated under stirring, 55 DEG C is cooled to, then adds 5- hydroxybenzimidazole 40.4g, stir lower heat Backflow, react 20~24h.
Ethyl acetate 100ml is added into reaction bulb, 0.5h dissolvings are stirred at 30~50 DEG C, are then reduced to -5 DEG C, are added dropwise just Hexane 30ml, stirs 1h, and suction filtration obtains light yellow solid, 40 DEG C of dryings to constant weight, obtains 56.41 grams, yield 85.8%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl }-piperazine The synthesis of pyridine -4- bases-t-butyl carbamate (IV)
II (50 grams), III (30.14 grams), potassium carbonate 80g, DIPHOS 4.3g, toluene 700ml, are added in 2L three-necked bottles, add Enter acid chloride 0.9g, stir.Stirring is lower to heat up, and temperature control reacts 24~30h at 80~100 DEG C.After the completion of reaction, it is cooled to 55 DEG C add dichloroethanes 700ml.10min is stirred, adds the solid in diatomite filtering reacting liquid, the filter cake chloroethenes of 500ml bis- Alkane rinses.Concentrate the filtrate to it is dry, add ethyl acetate 480ml, be heated to flowing back, be cooled to 20~25 DEG C of 10~20h of crystallization. The solid separated out is filtered, 50 DEG C of forced air dryings, obtains white solid, the amount of obtaining 70.51g, yield 93.90%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl } -4- The synthesis of amino piperidine (V)
By compound (1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline -8- Base }-piperidin-4-yl-t-butyl carbamate 5g, caustic alcohol 2.8g, 2- methyltetrahydrofuran 30ml and water 0.08ml be added to In 100ml three-necked bottles, stir.The mixture is heated to flowing back, and stirs 3~4h under reflux.
TLC is detected, and reaction is complete.Stop heating, add purified water 60ml, extracting and demixing.Aqueous phase is extracted with 2 × 20ml of ethyl acetate Take, merge organic phase, washed with saturated nacl aqueous solution 20ml.Be concentrated under reduced pressure organic phase, and 30ml is added into condensate residue Ethyl acetate, in 20~25 DEG C of stirring and crystallizing 6h.The solid separated out is filtered, filtrate decompression is concentrated to dryness.Added into residue 24ml ethyl acetate, in 20~25 DEG C of 10~12h of stirring and crystallizing.Filter the solid separated out, dry white solid product, the amount of obtaining 3.68g, yield 90.3%.
1H NMR test (referring to accompanying drawing)
(d6-DMSO):δ 9.176 (s, 1H), δ 8.88-8.91 (d, 1H, J=8.7Hz), δ 8.51-8.53 (d, 1H, J= 9.0Hz), δ 8.13-8.15 (d, 1H, J=9.0Hz), δ 7.6 (d, 1H, J=7.5Hz), 7.49 (t, 1H, J=7.9Hz) 7.39 (d, 1H, J=2.4Hz), 7.29 (d, 1H, J=7.6Hz), 7.19 (dd, 1H, J=9.2hz, 2.5Hz) 4.56 (d, 2H, J= 5.6Hz), 4.34 (d, 2H, J=5.7Hz), 4.14 (s, 2H), 3.74 (d, 2H, J=10.1Hz), 2.77 (m, 3H), 1.91 (d, 2H, J=11.1Hz), 1.68 (m, 2H), 1.41 (s, 3H)



SET 2




///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Background
Type III Receptor tyrosine kinase, including FLT3, PDGFRα and PDGFRβ, have been directly implicated in the pathogenesis of epithelial, mesenchymal, and hematological malignancies.[6]
Mutations of FLT3 comprise one of the most frequently identified types of genetic alterations in Angiomyolipoma.[7][8] Approximately one-third of AML patients present with a mutation in this gene.[9] The majority of these mutations result in constitutive activation of downstream signaling pathways and aberrant cell growth.[7] Mutations in FLT3 have also been reported in acute lymphoblastic leukemia (ALL)[10] and myelodysplastic syndrome (MDS).[11]
Activating mutations in PDGFRA have been detected in 5-12% of Gastrointestinal stromal tumor.[12] Fusion of PDGFRA has been found to be responsible for hematological malignances like hypereosinophilic syndrome.[13] The amplification of chromosome 4q12, the site of the PDGFRA gene[citation needed], has been identified in 13-29% of adult gliomas[citation needed] and in 29% to 36% of diffuse intrinsic pontine gliomas (DIPG)[citation needed], a subset of high-grade gliomas (HGG) in pediatric patients. Activation of PDGFRB, a third member of the type III RTK family, has been implicated in the development of chronic myelomonocytic leukemia due to the fusion of PDGFRB with the TEL gene.[13] Furthermore, PDGFB translocation to the COL1A1 gene locus has been identified to be responsible for dermatofibrosarcoma protuberans (DFSP).[13] In cancer cells, PDGFR promotes tumor development and migration via proto-oncogenic downstream mediators like AKT and MEK[citation needed]. In stromal fibroblasts, PDGFRα activation leads to local tissue invasion, production and secretion of VEGF, and elevated intratumoral interstitial pressure[citation needed]. In stromal pericytes, PDGFRβ activation mediates vascular stability.[13] Thus, either FLT3 or PDGF/PDGFR pathway is the primary driver of oncogenesis in the above malignancies and can be targeted by crenolanib therapy[citation needed].
Mechanism
FLT3: wild-type and mutant
Crenolanib inhibits both wild type FLT3 and its constitutively active mutations. In vitro studies have shown that crenolanib has low Kd for the FLT3 enzyme with constitutively activating internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations, D835H and D835Y, as compared to wild type. Crenolanib tightly binds to FLT3-ITD, FLT3-D835H and FLT3-D835Y with Kd of 0.74 nM, 0.4 nM, and 0.18 nM, respectively.[14] Crenolanib inhibits the phosphorylation of the FLT3-ITD receptor in transfected TF-1 cells and the FLT3-D835Y TKD mutation in transfected Ba/F3 cells at nanomolar IC50 concentrations of 1.3 nM and 8.8 nM, respectively.[15] Immunoblot experiments performed in the Molm14 FLT3-ITD positive cell line show that crenolanib inhibits downstream signaling of FLT3 at a concentration of 10 nM.[15] MTT assay measurements of crenolanib cytotoxicity evaluated in the FLT3-ITD expressing cell lines Molm14 and MV411, showed that crenolanib is toxic at IC50 concentrations of 7 nM and 8 nM, respectively.[15]
PDGFRα: wild-type and mutant
Crenolanib has been shown to inhibit PDGFRα with an IC50 of 0.4 ng/mL in porcine aortic epithelial cell lines. In Chinese hamster ovary (CHO) cells expressing PDGFRα, crenolanib inhibited the phosphorylation of wild type PDGFRα at an IC50 of 10 nM.[16] Additionally, crenolanib completely blocked PDGFRα phosphorylation and downstream AKT signaling at a concentration between 0.1 and 1 uM in Ink4a/Arf-/- mouse astrocytes transfected to stably co-express both human PDGFRα and PDGF AA.[17] The lung cancer cell line H1703, which is reported to have amplification of both PDGFRA (4q12) and PDGFC (4q32) genes on chromosome 4, and also overexpress PDGFRα, was sensitive to crenolanib with an IC50 of ~80 nM.[18] In CHO cells expressing an activating exon 18 (D842V) PDGFRα mutation, crenolanib was effective at an IC50 of 6nM and IC90 of 25nM. In addition, crenolanib also inhibited phosphorylation of the double mutants PDGFRα (V561D + D842V and T674I + D842V).[16]
PDGFRβ: wild-type
Crenolanib has been shown to inhibit PDGFRβ with an IC50 of 0.8 ng/mL in porcine aortic epithelial cell lines. Crenolanib inhibits the ability of recombinant PDGFRβ to phosphorylate a synthetic tyrosine substrate (poly-glutamic acid-tyrosine), with an IC50 of 0.4 ng/mL. Evaluation of the antitumor activity of crenolanib in a genetically engineered BSG DIPG mouse model showed that it is highly selective for PDGFRβ with an IC50 of 10 nM when measured by BrdU assay and 1.25 uM by MTT assay.
C-Kit: wild-type and mutant
Crenolanib has been shown to have IC50 and Kd values of 67 nM and 78 nM, respectively, for wild type c-KIT in in vitro assays[citation needed]. Similar assays show that crenolanib inhibits c-KIT activating mutations D816H and D816V with IC50 concentrations of 5.4 and 2.5 nM, respectively.[14][citation needed] Human bone marrow progenitor cell growth assays showed that crenolanib has modest effects on GM-CSF and BFUE driven colony formation at the IC50 concentration of 20 nM.[15]
Clinical
Phase I single-agent[19] and Phase Ib combination[20] studies have investigated the clinical pharmacology of crenolanib in patients with cancer. Pharmacokinetic and safety studies of Crenolanib administered alone or in combination with docetaxel with or without axitinib have been completed. Results suggest that Crenolanib is well tolerated as a single agent, and can also be safely combined with docetaxel and axitinib due to their non-overlapping toxicity profiles.
Clinical trials
- Clinical trial number NCT01229644 for “A Phase II Study of Crenolanib (CP-868,596), a Selective and Potent Inhibitor of PDGFR, for the Treatment of Adult Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT01243346 for “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene” at ClinicalTrials.gov
- Clinical trial number NCT01393912 for “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma” at ClinicalTrials.gov
- Clinical trial number NCT01522469 for “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations” at ClinicalTrials.gov
- Clinical trial number NCT01657682 for “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations” at ClinicalTrials.gov
References
- ^ “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ A. Ramachandran; H. Marshall; V. Jain. “CRENOLANIB, A NOVEL TYPE I, MUTANT -SPECIFIC INHIBITOR OF CLASS III RECEPTOR TYROSINE KINASES, PREFERENTIALLY BINDS TO PHOSPHORYLATED KINASES” (PDF). gistsupport.org. Retrieved 2014-04-08.
- ^ Lemmon, Mark A.; Schlessinger, Joseph (2010). “Cell Signaling by Receptor Tyrosine Kinases”. Cell. 141 (7): 1117–34. doi:10.1016/j.cell.2010.06.011. PMC 2914105. PMID 20602996.
- ^ Jump up to:a b Takahashi, S (2011-04-01). “Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications”. J Hematol Oncol. 4: 13. doi:10.1186/1756-8722-4-13. PMC 3076284. PMID 21453545.
- ^ Cancer Genome Atlas Research Network; Ley, T. J.; Miller, C.; Ding, L.; Raphael, B. J.; Mungall, A. J.; Robertson, A.; Hoadley, K.; Triche Jr, T. J.; Laird, P. W.; Baty, J. D.; Fulton, L. L.; Fulton, R.; Heath, S. E.; Kalicki-Veizer, J.; Kandoth, C.; Klco, J. M.; Koboldt, D. C.; Kanchi, K. L.; Kulkarni, S.; Lamprecht, T. L.; Larson, D. E.; Lin, L.; Lu, C.; McLellan, M. D.; McMichael, J. F.; Payton, J.; Schmidt, H.; Spencer, D. H.; et al. (2013). “Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia”. New England Journal of Medicine. 368 (22): 2059–2074. doi:10.1056/NEJMoa1301689. ISSN 0028-4793. PMC 3767041. PMID 23634996.
- ^ “The Impact of FLT3 Mutations on the Development of Acute Myeloid Leukemias”. Hindawi.com. Retrieved 2014-04-08.
- ^ Xu, F; Taki, T; Yang, HW; Hanada, R; Hongo, T; Ohnishi, H; Kobayashi, M; Bessho, F; Yanagisawa, M; Hayashi, Y (2014-01-24). “Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children”. Br. J. Haematol. 105 (1): 155–62. doi:10.1111/j.1365-2141.1999.01284.x. PMID 10233379. S2CID 40898615.
- ^ Yokota, S; Kiyoi, H; Nakao, M; Iwai, T; Misawa, S; Okuda, T; Sonoda, Y; Abe, T; Kahsima, K; Matsuo, Y; Naoe, T (2014-01-24). “Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines”. Leukemia. 11 (10): 1605–9. doi:10.1038/sj.leu.2400812. PMID 9324277.
- ^ Heinrich, M. C.; Corless, CL; Duensing, A; McGreevey, L; Chen, CJ; Joseph, N; Singer, S; Griffith, DJ; Haley, A; Town, A; Demetri, GD; Fletcher, CD; Fletcher, JA (2003). “PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors”. Science. 299 (5607): 708–10. doi:10.1126/science.1079666. PMID 12522257. S2CID 11725958.
- ^ Jump up to:a b c d Östman, Arne; Heldin, Carl‐Henrik (2007). PDGF Receptors as Targets in Tumor Treatment. Advances in Cancer Research. Vol. 97. pp. 247–274. doi:10.1016/S0065-230X(06)97011-0. ISBN 9780120066971. PMID 17419949.
- ^ Jump up to:a b Muralidhara, C.; Ramachandran, A.; Jain, V. K. (2012). “Abstract 3683: Crenolanib, a novel Type I, mutant-specific inhibitor of Class III receptor tyrosine kinases, preferentially binds to phosphorylated kinases”. Cancer Research. 72 (8 Supplement): 3683. doi:10.1158/1538-7445.AM2012-3683.
- ^ Jump up to:a b c d Galanis, A.; Rajkhowa, T.; Muralidhara, C.; Ramachandran, A.; Levis, M. (2012). “Abstract 3660: Crenolanib: A next generation FLT3 inhibitor”. Cancer Research. 72 (8 Supplement): 3660. doi:10.1158/1538-7445.am2012-3660.
- ^ Jump up to:a b Heinrich, M. C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. (2012). “Crenolanib Inhibits the Drug-Resistant PDGFRA D842V Mutation Associated with Imatinib-Resistant Gastrointestinal Stromal Tumors”. Clinical Cancer Research. 18 (16): 4375–84. doi:10.1158/1078-0432.CCR-12-0625. PMID 22745105.
- ^ Yang, X.-L.; Mashimo, T.; Su, Y.; Vemireddy, V.; Guntipalli, P.; Ramachandran, A.; Chaudhary, P.; Mickey, B.; Hatanpaa, K.; Maher, E.; Bachoo, R. M. (2011). “Abstract 1111: Preclinical evaluation of CP868,596, a novel PDGFR Inhibitor for treatment of glioblastoma”. Cancer Research. 71 (8 Supplement): 1111. doi:10.1158/1538-7445.am2011-1111.
- ^ Peyton, M.; Chaudhary, P.; Ramachandran, A.; Minna, J. (2011). “Abstract 3601: CP-868,596, a highly potent and selective PDGFR TKI inhibits growth of PDGFR -driven lung cancer cells”. Cancer Research. 71 (8 Supplement): 3601. doi:10.1158/1538-7445.am2011-3601.
- ^ Lewis, N. L.; Lewis, L. D.; Eder, J. P.; Reddy, N. J.; Guo, F.; Pierce, K. J.; Olszanski, A. J.; Cohen, R. B. (2009). “Phase I Study of the Safety, Tolerability, and Pharmacokinetics of Oral CP-868,596, a Highly Specific Platelet-Derived Growth Factor Receptor Tyrosine Kinase Inhibitor in Patients with Advanced Cancers”. Journal of Clinical Oncology. 27 (31): 5262–9. doi:10.1200/jco.2009.21.8487. PMC 2773478. PMID 19738123.
- ^ Michael, M; Vlahovic, G; Khamly, K; Pierce, K J; Guo, F; Olszanski, A J (2010). “Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor”. British Journal of Cancer. 103 (10): 1554–61. doi:10.1038/sj.bjc.6605941. PMC 2990584. PMID 20959830.
External links
- “PDGFR Inhibitor CP-868596 (Code C64639)”, National Cancer Institute Thesaurus.
- “PDGFR and Human Cancer” , AROG Pharmaceuticals LLC.
| Names | |
|---|---|
| IUPAC name1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine | |
| Other namesCP-868,596; AR-868,596-26 | |
| Identifiers | |
| CAS Number | 670220-88-9 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:145365 |
| ChEMBL | ChEMBL2105728 ChEMBL2146086 |
| ChemSpider | 8541584 |
| IUPHAR/BPS | 7882 |
| KEGG | D10102 |
| PubChemCID | 10366136 |
| UNII | LQF7I567TQ |
| CompTox Dashboard (EPA) | DTXSID50985873 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C26H29N5O2 |
| Molar mass | 443.551 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
 verify (what is ?) verify (what is ?) | |
| Infobox references |
//////////Crenolanib, UNII-LQF7I567TQ, креноланиб , كرينولانيب , 克拉尼布, CP-868,596-26, AR-868,596-26
CC1(COc2ccc3c(c2)ncn3c4ccc5cccc(N6CCC(N)CC6)c5n4)COC1.OS(=O)(=O)c7ccccc7
