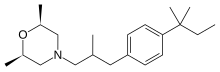
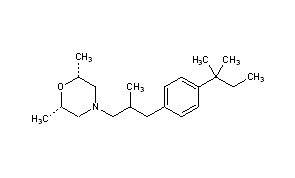
AMOROLFINE(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methyl-2-butanyl)phenyl]propyl}morpholine
(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methylbutan-2-yl)phenyl]propyl}morpholine
78613-35-1[RN]
(±)-cis-2,6-Dimethyl-4-(2-methyl-3-(p-tert-pentylphenyl)propyl)morpholine
Ro 14-4767-002
аморолфин , أمورولفين ,阿莫罗芬 ,
Title: Amorolfine
CAS Registry Number: 78613-35-1
CAS Name:cis-4-[3-[4-(1,1-Dimethylpropyl)phenyl]-2-methylpropyl]-2,6-dimethylmorpholine
Additional Names:cis-4-[3-(4-tert-amylphenyl)-2-methylpropyl]-2,6-dimethylmorpholine; (±)-cis-2,6-dimethyl-4-[2-methyl-3-(p-tert-pentylphenyl)propyl]morpholine
Manufacturers’ Codes: Ro-14-4767/000
Molecular Formula: C21H35NO
Molecular Weight: 317.51
Percent Composition: C 79.44%, H 11.11%, N 4.41%, O 5.04%
Literature References: Antimycotic morpholine derivative; inhibits fungal ergosterol biosynthesis. Prepn (unspec stereochem): A. Pfiffner, K. Bohnen, DE2752096; A. Pfiffner, US4202894 (1978, 1980 both to Hoffmann-La Roche); of cis-form: NL8004537 (1980 to Hoffmann-La Roche). In vitro comparative antifungal spectrum: S. Shadomy et al.,Sabouraudia22, 7 (1984). Mechanism of action: A. Polak-Wyss et al.,ibid.23, 433 (1985); A. Polak, Ann. N.Y. Acad. Sci.544, 221 (1988). LC determn in pharmaceutical formulations: M. A. Czech et al.,J. Pharm. Biomed. Anal.9, 1019 (1991). Series of articles on mode of action and clinical trials: Clin. Exp. Dermatol.17, Suppl. 1, 1-70 (1992). Review of pharmacology and clinical efficacy: M. Haria, H. M. Bryson, Drugs49, 103-120 (1995).
Properties: bp0.1 120°.
Boiling point: bp0.1 120°
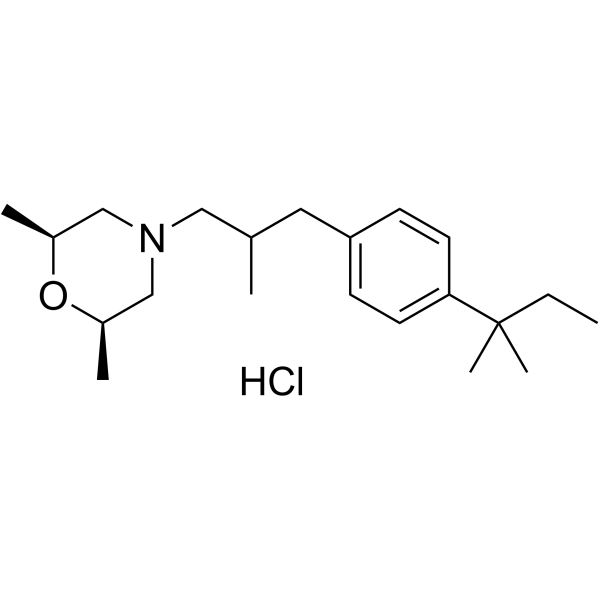
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent.
Derivative Type: Hydrochloride
CAS Registry Number: 78613-38-4
Manufacturers’ Codes: Ro-14-4767/002
Trademarks: Loceryl (Roche)
Molecular Formula: C21H35NO.HCl
Molecular Weight: 353.97
Percent Composition: C 71.26%, H 10.25%, N 3.96%, O 4.52%, Cl 10.02%
Therap-Cat: Antifungal (topical).
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent. Target: Antifungal Amorolfine is an antifungal showing activity against fungi pathogenic to plants, animals and humans. Amorolfine possesses a broad antifungal spectrum including dermatophytes, yeasts, dimorphic fungi and moulds and is not only fungistatic but fungicidal against most species [1]. At 0.2, 2 and 5 micrograms/ml amorolfine did not have any significant inhibitory or enhancing effect on phagocytosis whether following simultaneous addition of blastospores and drug to the neutrophils, prior treatment of neutrophils for 2 h before addition of blastospores or prior treatment of blastospores for 2 h. Simultaneous addition of amorolfine resulted in a significant increase in killing at all concentrations. This increase was not significantly enhanced by either preincubation of neutrophils or blastospores for 2 h with the drug [2].
Amorolfine (or amorolfin), is a morpholineantifungal drug that inhibits Δ14-sterol reductase and cholestenol Δ-isomerase, which depletes ergosterol and causes ignosterol to accumulate in the fungal cytoplasmiccell membranes. Marketed as Curanail, Loceryl, Locetar, and Odenil, amorolfine is commonly available in the form of a nail lacquer, containing 5% amorolfine hydrochloride as the active ingredient. It is used to treat onychomycosis (fungal infection of the toe- and fingernails). Amorolfine 5% nail lacquer in once-weekly or twice-weekly applications has been shown in two studies to be between 60% and 71% effective in treating toenail onychomycosis; complete cure rates three months after stopping treatment (after six months of treatment) were 38% and 46%. However, full experimental details of these trials were not available and since they were first reported in 1992 there have been no subsequent trials.[1]
It is a topical solution for the treatment of toenail infections.[2][3] Systemic treatments may be considered more effective.[1]
It is approved for sale over-the-counter in Australia, Brazil, Russia, Germany and the UK, and is approved for the treatment of toenail fungus by prescription in other countries. It is not approved for the treatment of onychomycosis in the United States or Canada, but can be ordered from there by mail from other countries.[4]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Indian Pat. Appl., 2010MU01980,







SYN
https://pubs.rsc.org/en/content/articlelanding/2017/ob/c6ob02765b/unauth
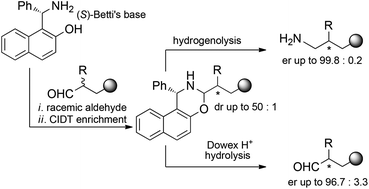
The acid-promoted crystallization-induced diastereoisomer transformation (CIDT) of naphthoxazines derived from racemic O-protected 2-substituted 4-hydroxybutyraldehydes and enantiopure Betti’s base allows the deracemization of the starting aldehydes with ee up to 96%. As an alternative, reduction with lithium aluminum hydride of the diastereoisomerically enriched naphthoxazines leads to enantioenriched primary amines. The utility of the latter strategy was demonstrated by applying it to the synthesis of enantioenriched fenpropimorph and to the first synthesis of enantiopure amorolfine, with ee up to 99.5%.
PATENT
https://patents.google.com/patent/WO2013097629A1/en
Amorolfine hydrochloride, chemical name is cis-4-[3-[4-(1,1-dimethyl-propyl)phenyl]-2-mercaptopropyl]-2 , 6-diamidino-morpholine hydrochloride, CAS registration number is 78613-38-4, the chemical knot is as follows:

Amoxifen hydrochloride is an antifungal drug developed by Roche and launched in 1991 under the trade name Leceryl. Regarding the synthesis process of amorolfine hydrochloride, the prior art has been described:


US7795425B2 synthetic route: (1) 2-nonyl cinnamaldehyde is condensed with cis-2,6-dimethylmorpholine to give cis-4-(3-phenyl-2-methylpropyl)-2,6- Dimercapto-morpholine hydrochloride, (2) cis-4-(3-phenyl-2-methylpropyl)-2,6-dimethyl-morpholine hydrochloride followed by 2-methyl – 2-chlorobutane, with acid Catalytic, Heck reaction occurs, and amorolfine is obtained. In step (1), palladium carbon catalytic hydrogenation is required, so the cost is high; in addition, there may be multiple rearrangement reactions in step (2), many by-products, difficult product purification, low quality of finished product and low yield. And it requires a low temperature reaction equipment of -40 ~ -65 °C, which consumes a lot of energy. International patent application WO2007113218A1 improves the synthesis method of amorolfine hydrochloride, the first step of Heck reaction, 4-iodo-t-amylbenzene and 2-methylallyl alcohol are reacted in the presence of a palladium catalyst and a base to obtain 3-un Butyl phenyl-2-methylpropanal, the reaction solvent is selected from N,N-dimercaptocarboxamide (abbreviated as DMF), polar protic solvent or non-polar solvent; second step reductive amination reaction, 3 – tert-amylphenyl-2-mercaptopropanal is reacted with cis-2,6-dimercaptomorpholine to give amorolfine, the reducing agent is selected from palladium

The WO2007113218A1 process still has defects: (1) The first step of the Heck reaction, the reaction solvent DMF is moderately toxic, and the International Agency for Research on Cancer (IARC) considers it to be a carcinogen. DMF is chemically stable and can exist for a long time in wastewater. It is highly polluted by water and difficult to biodegrade. Its BOD5/COD value is 0.065 ( BOD5/COD is an indicator of biodegradability of wastewater, and 0.3 is the lower limit of biodegradable degradation of wastewater). value). Wastewater treatment costs are high during large production. Although the boiling point of DMF is 154 ° C, it is unstable under alkaline conditions, especially at high temperatures, and decomposition starts at 100 ° C or higher. The polar protic solvent, such as the lower alcohol described in the patent, cannot meet the high temperature reaction requirements, and the high boiling polar protic solvent has poor solubility to the catalyst and is difficult to react. The non-polar solvent does not substantially dissolve the palladium catalyst, so the application value is not large. (2) The second step of reductive amination reaction, using expensive The cost of catalytic hydrogenation of heavy metal palladium is high, and the high pressure reaction equipment is unsafe; the reduction of metal borohydride is easy to generate a large amount of hydrogen, which poses a safety hazard, and also reduces 3-tert-pentylphenyl-2-methylpropanal to The corresponding alcohol increases the impurities; the reduction by-product of the metal cyanoborohydride is highly toxic. (3) The product yield was low, and the total yield of the product of the example was about 50%. None of the purity of the products and intermediates has been disclosed.The chemical reaction equation of the present invention is expressed as follows:

(la) (lb)In a 10L clean reaction kettle, add 2600 mL of acetic anhydride, 5200 mL of glacial acetic acid, 350 g of sodium periodate, break 1236 g, cool to 5 ° C, add 810 mL of sulfuric acid, control the dropwise addition within 1 hour, and then add 1130 g of t-amyl. The benzene was stirred at room temperature for more than 16 hours, and the reaction of the raw materials was confirmed by thin layer chromatography. The reaction mixture was poured into a mixture of 8 L of water and 4 L of dichloromethane, and the mixture was separated. The organic layer was washed with 4L of 25% aqueous sodium sulfite, and the organic layer was dried over anhydrous sodium sulfate. It was 4-iodo-t-amylbenzene 2013 g, yield: 96%, and the GC purity was 94.2%. NMR spectral data: (400 MHz, CDC1 3 ): 0.73 (3H, t, J = 7.4 Hz), 1.31 (6H, s), 1.67 (2H, q, J – 7.4 Hz), 7.13 (2H, d, J = 8.56 Hz), 7.66 (2H, d, J = 8.56 Hz) 0 Example 22 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, and 300 g of palladium acetate and 1.7 kg of sodium hydrogencarbonate were added. Finally, 2.5 kg of 2-mercaptopropanol was added, the temperature was raised to 105 C, and the GC content of 4-iodo-t-amylbenzene was measured to monitor the progress of the reaction, and the reaction was completed for 2 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, wash with 20 L of water, rectify the organic phase, collect 125-128 ° C fraction (vacuum degree ≤ -0.099)\3⁄4^), and obtain 3- Tert-amylphenyl-2-mercaptopropanal L41 kg, yield: 88.6%, GC purity: 93.5%. NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J =7.43 Hz), 2.60 (13⁄4 dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9,75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of ethyl acetate was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 600 g of 2,6-dimethylmorpholine was added dropwise. , add about 30 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 C, the addition was completed, and the temperature was raised to 18 ° C for 30 minutes. After cooling to 10 Torr, 1,3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 18 ° C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. Ended in 2 hours. After cooling to 10 ° C or lower, the pH was adjusted to 10 with a sodium hydroxide solution, and the layers were allowed to stand, and the organic layer was washed with 4 L of water. The organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 V for 14 hours to obtain 1.59 kg of amorolamine hydrochloride, yield: 85.6%, HPLC purity: 99.6%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J=7, 2Hz), 1.03 (3H, d, J=6.8Hz), 1.15(6H, d, J=6 , 0 Hz), 1.25 (63⁄4 s), 1.64 (2H, m, J = 7.6 Hz), 2.34 (1H, d, J = 6.8 Hz), 2.48 (23⁄4 d, J = 6.8 Hz), 2.75 (2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz) 5 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 3 In a 10 L clean reaction kettle, 2 kg of 4-substituted tert-amylbenzene prepared according to the method of Example 1 and 6 L of N-mercaptopyrrolidone were protected by nitrogen, stirring was started, and 150 g of palladium acetate and 2.5 kg of dipotassium hydrogen phosphate were added. Finally, 1.8 kg of 2-methylallyl alcohol was added, and the temperature was raised to 130. C reaction, the GC content of 4-deuterated tert-amylbenzene was measured to control the progress of the reaction, and the reaction was completed for 10 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, dissolve 20 L of water, concentrate the organic phase, recover ethyl acetate, and add the residue to 10 L of saturated sodium hydrogen sulfite solution at room temperature to precipitate solid. The mixture was stirred for 6 hours, filtered, and filtered, washed with EtOAc EtOAc EtOAc EtOAc. The filtrate was concentrated to dry ethyl acetate to give 1. <RTI ID=0.0>#</RTI><RTIgt;</RTI><RTIgt;</RTI><RTIgt; -NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J-7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of ethyl acetate in a 10 L reactor, protect with nitrogen, cool to 10 C, and add 1.2 kg of 2,6-dimethylmorpholine dropwise. , 40 minutes added. Then, 780 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 ° C, 2.3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 25 ° C, and the GC content of 3-tert-amylpyridyl-2-methylpropanal was detected to monitor the progress of the reaction. The reaction was completed in 2 hours. Cool to below 10 ,, adjust the pH to 11 with sodium hydroxide solution, let stand for stratification, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 70 ° C decompression After drying for 14 hours, 1.75 kg of amorolfine hydrochloride was obtained, yield: 84.6%, HPLC purity: 99.7%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2Hz), 1.03 (3H, d, J = 6.8Hz), L15(6H, d, J=6.0Hz ), 1.25(6H, s), L64(2H 5 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H 5 d, J=11.2Hz), 3·9(2Η, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H ; dd, J = 8.4 Hz). Example 4In a 10 L clean reaction kettle, 2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1, 2 N of N-methylpyrrolidone, protected by nitrogen, stirring was started, and palladium nitrate 6 g, acetic acid was added. Sodium 627 g, and finally 592 g of 2-methylallyl alcohol was added thereto, and the temperature was raised to 140 ° C to carry out a reaction. The GC content of 4-deactivated t-amylbenzene was examined to monitor the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 8 L of ethyl acetate, dissolve in 16 L of water, rectify the organic phase, collect 125-128 C fraction (vacuum degree ≤ -0.0991 ^ & ) to give 3-tert-pentylphenyl 2-mercaptopropanal 1.37 kg, yield: 86%, GC purity: 93.0%. MR spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1 , 11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , 3=1 A3 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).The above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5 L of dichloromethane was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 1.6 kg of 2,6-dimethyl was added dropwise. Morpholine, added in 45 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 Torr for 60 minutes. After cooling to 10 ° C, 1.6 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. The end of the hour. Cool to below 10 °C, adjust the pH to 10 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4L water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 °C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.6%, HPLC purity: 99.6%. iH-NMR spectral data: ! H NM (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J= 6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H , d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 52 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, 30 g of palladium chloride and 750 g of sodium hydrogencarbonate were added. Finally, 1.3 kg of 2-methylallyl alcohol was added, and the mixture was heated to 120 ° C to measure the GC content of 4-iodo-t-amylbenzene to control the progress of the reaction, and the reaction was completed for 13 hours. It was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was dissolved in 8 L of chloroform, washed with 16 L of water, and the organic phase was concentrated. The ethyl acetate was recovered. The residue was added dropwise to 10 L of saturated sodium hydrogensulfite solution at room temperature to precipitate a solid. Hour, filter, filter cake washed with 5 L of ethyl acetate, solid dispersed in 3 L 3 mol / liter The mixture was stirred at room temperature for 5 hours, and the reaction mixture was dried over EtOAcjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Yield: 91,7%, GC purity: 98.8%. – Spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J=7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of absolute ethanol in a 10 L reactor, protect with nitrogen, cool to 10 ° C, and add 600 g of 2,6-dimercaptomorpholine. , added in 30 minutes. Then, 500 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was monitored at 10 Torr, and the GC content of 3-tert-pentylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction. The end of the hour. 10. Under C, adjust the pH value to 11 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 7 CTC minus After drying for 14 hours, 1.45% of amorolfine hydrochloride was obtained, yield: 87.0%, HPLC purity: 99.7% – NMR spectral data: J H NMR (400 MHz 5 CD 3 OD) 6: 0.64 (3H, t, J= 7,2Hz), 1.03(3H, d, J=6.8Hz), 1.15(6H, d, J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34( 1H ? d, J = 6.8 Hz), 2.48 (2H, d, J = 6.8 Hz), 2.75 (23⁄4 d, J = 6.0 Hz), 3.1 (2H, d, J = 8.8 Hz), 3.4 (2H, d , J = 11.2 Hz) 5 3.9 (2H, m), 7.16 (2H, dd, J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 62 kg of 4-iodo-t-amylbenzene prepared in accordance with the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel. The mixture was stirred under nitrogen, stirring was started, 10 g of palladium acetate was added, and 800 g of carbonic acid was added. 1.1 kg of 2-mercaptopropanol was heated to 80 ° C, and the GC content of 4-deactivated t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add 8 L of chloroform to dissolve, 16 L of water, rectify the organic phase, collect 125-128 ° C 真空 (vacuum degree ≤ -0.099 ^ ^ & ), to obtain 3-tert-amylbenzene Base-2-mercaptopropanal 1.42 kg, yield: 89.2%, GC purity: 92.5%. ^- MR Spectral Data: (400 MHz, CDC1 3 ): 0.69 (33⁄4 t, J=7.45 Hz), 1.11 (3H, d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of decyl alcohol was added to a 10 L reactor, protected with nitrogen, cooled to 10 C, and 600 g of 2,6-dimethylmorpholine was added dropwise for 30 minutes. Plus finished. Then, 500 mL of water acetic acid was added dropwise, the temperature was kept at 10 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. End of 2 hours. Cool to 10 ° C, adjust the pH to 10 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 1.5, filter, filter The cake was dried under reduced pressure at 65 C for 15 hours to obtain 1.46 kg of amorolfine hydrochloride, yield: 90.1%, HPLC purity: 99,8%. ^-NMR spectral data: l R NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), U5 (6H, d, J = 6.0Hz), 1.25(6H, s), 1.64(23⁄4 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=l 1.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 72 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-decylpyrrolidone were added to a 10 L clean reaction kettle, protected by nitrogen, stirring was started, and 75 g of palladium acetate and 2.0 kg of disodium hydrogen phosphate were added. Finally, 780 g of 2-methylallyl alcohol was added, and the temperature was raised to 125 Torr. The GC content of 4-iodo-t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 8 hours. The mixture was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was evaporated, evaporated, evaporated, evaporated, evaporated. The solid was precipitated, stirred for 6 hours, filtered, and the filter cake was washed with 5 L of ethyl acetate. The solid was dispersed in 10 L 2 mol/L hydrochloric acid, stirred at room temperature for 5 hours, and the reaction mixture was extracted with 10 L of ethyl acetate. The mixture was dried, filtered, and the filtrate was evaporated to ethyl acetate to ethylamine (ethyldiethyldithioacetate). 3⁄4-NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add the above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5L hydrazine, in a 10L reactor Under nitrogen atmosphere, cooled to 10 Torr, 700 g of 2,6-dimercaptomorpholine was added dropwise, then 280 mL of glacial acetic acid was added, the temperature was maintained at 15 C, and then the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.0 kg of sodium triacetoxyborohydride was added, and 20 was added. The temperature was maintained under C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was examined to monitor the progress of the reaction, and the reaction was completed for 3 hours. Cool to below 10 ° C, adjust the pH to 11 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 ° C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.8%, HPLC purity: 99.6%. ^-NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H ; d, J = 6.0 Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) } 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz) , 7.27 (2H, dd, J = 8.4 Hz). Example 83-tert-pentylphenyl-2-mercaptopropanol lkg, 5 L of dichloromethane prepared by the method of Example 5 was added to a 10 L reactor, and was purged with nitrogen and cooled to 10. C, 1000 g of 2,6-dimethylmorpholine was added dropwise, then 400 mL of water acetic acid was added, the temperature was maintained at 15 ° C, and then the temperature was raised to 20 ° C for 60 minutes. After cooling to 0 C, 1.5 kg of sodium triacetoxyborohydride was added, and 6 C was added after the addition, and the GC content of 3-tert-pentylphenyl-2-mercaptopropanal was detected to monitor the progress of the reaction for 5 hours. End. Adjust the pH to 10 with sodium hydroxide solution at 6 °C, let stand for layering, wash the organic layer with 4L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 2, filter, filter cake and dry at 65 Ό for 14 hours under reduced pressure. , Amofufen hydrochloride 1.48kg, yield: 91.2%, HPLC purity: 99.7%. ^- MR spectral data: NMR (400MHz, CD 3 OD) 5: 0·64 (3Η, ΐ, J=7, 2Hz), 1.03(3Η, d, J=6.8Hz), 1.15(6H, d, J =6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75( 2H, d, J=6.0Hz), 3,1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(23⁄4 m), 7.16(2H, dd, J- 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 9Add 3-tert-pentylphenyl-2-mercaptopropanol lkg prepared in the same manner as in Example 2, 4 L of tetrahydrofuran, protect with nitrogen, cool to 10 ° C, add 820 g of 2,6-two Mercaptomorpholine, Then, 380 mL of glacial acetic acid was added, the temperature was maintained at 15 ° C, and then kept at room temperature for 60 minutes. After cooling to 10 ° C, 1.8 kg of sodium triacetoxyborohydride was added, and after 10 liters of the addition, the GC content of 3-tert-amylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction for 5 hours. End. The pH was adjusted to 10 with sodium hydroxide solution at 10 ° C, and the layers were allowed to stand. The organic layer was washed with 4 L of water, and the organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 Torr for 14 hours. , amlofol hydrochloride 1.41 kg, yield: 87.1%, HPLC purity: 99.8%. NMR spectral data: J H NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), L03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) ), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J-6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 1In a 1000 mL four-necked flask, 137 g of 4-deuterated tert-amylbenzene prepared according to the method of Example 1, 1.12 g of palladium acetate, 50.4 g of sodium hydrogencarbonate, N,N-dimethylformamide 500 mL, nitrogen gas, added 54 g of 2-mercaptopropanol, warmed to 10 (TC for 10 hours, cooled to room temperature, filtered, filter cake washed with hydrazine, hydrazine-dimethylformamide 300 mL, combined filtrate, poured into 2000 mL of saturated brine and 1000 mL The mixture was extracted with ethyl acetate, and the organic phase was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated, dried, and evaporated, and the residue was distilled in vacuo to collect fractions of 125-128 ° C (vacuum degree <-0.099 MPa) to obtain 3-un Amyl phenyl-2-mercaptopropanal 84 g, Yield: 77%, GC purity: 88.0% – R spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz) , 1.11 (3H : d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2,60 (1H, dd, J=13.52 Hz), 2.69 (1H, J-7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).109 g of the above 3-tert-amylphenyl-2-mercaptopropanal and 500 mL of ethanol were placed in a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2,6-dimethylmorpholine were added. Stir at room temperature for 30 minutes, cool to -15 ° C, add 15.93 g of sodium borohydride in 1 hour. After the addition, warm to 0 C for 2 hours, adjust the pH to 12 with 25% sodium hydroxide solution. The mixture was extracted with 2000 mL of saturated brine and 1000 mL of ethyl acetate. The organic phase was washed with water and concentrated to dryness. The obtained residue was added to 500 mL of isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, and washed with isopropyl ether. , the filter cake is dried under reduced pressure at 70 ° C for 14 hours to obtain hydrochloric acid. Morofen 119 g, yield: 67%, HPLC purity: 97.1%. 3⁄4-NMR spectral data: ‘H NMR (400 MHz, CD 3 OD) 5: 0, 64 (3H, t, J = 7, 2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d , J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd , J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 2109 g of 3-tert-pentylphenyl-2-methylpropanal prepared according to the method of Comparative Example 1 and 500 mL of methanol were added to a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2, 6 were added. – dimethylmorpholine, stirred at room temperature for 30 minutes, cooled to -15 ° C, replaced with nitrogen, added 5 g of 0% palladium on carbon, passed through hydrogen, reduced at 40 ° C, 4 atm, until the hydrogen pressure did not decrease, The reaction is complete. Cool to room temperature, replace with nitrogen, filter, adjust the pH of the filtrate with 25% sodium hydroxide solution, add 2000 mL of saturated brine and 1000 mL of ethyl acetate for extraction, wash the organic phase, concentrate and dry, add the residue to 500 mL Isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, washed with isopropyl ether, and the filter cake was dried under reduced pressure at 70 ° C for 14 hours to obtain amolofol hydrochloride 113 g, yield: 64%. HPLC purity: 97.8%. NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) , 1.25(6H, s), 1.64(2H, m, J-7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) ? 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J-11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J=8, 4Hz).
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0447947A1 *1990-03-231991-09-25BASF AktiengesellschaftN-(3-Phenyl-2-methylpropyl and -methyl-prop-2-enyl)-azaheterocyclesWO2007113218A1 *2006-04-032007-10-11Galderma S.A.Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine)Family To Family CitationsEP1749826A1 *2005-07-282007-02-07Galderma S.A.Process of producing bepromolineCN101485625B *2009-02-192010-09-22中国药科大学Amoluofen emulsifiable paste
Publication numberPriority datePublication dateAssigneeTitle
CN105130808A *2015-08-132015-12-09上海瑞博化学有限公司High purity 2,5-dimethyl-3,4-dihydroxy methylbenzoate synthesis methodFamily To Family CitationsCN103288768B *2013-06-182015-02-18中国人民解放军第四军医大学Asymmetric synthetic method of optical pure amorolfine hydrochlorideCN104926629B *2015-05-302016-06-22江苏科本医药化学有限公司Domino reaction is utilized to prepare the green method of 3,3-diaryl acrylic aldehydeCN108997246B *2017-06-062021-08-31江苏礼华生物技术有限公司Preparation method of amorolfine hydrochlorideCN110498729A *2019-09-092019-11-26武汉诺安药业有限公司A kind of clean method for preparing of hydrochloric acid Amorolfine intermediate
Notes
- ^ Jump up to:a b Williams HC (2003). Evidence-Based Dermatology. Blackwell. ISBN 9781444300178.
- ^ Flagothier C, Piérard-Franchimont C, Piérard GE (March 2005). “New insights into the effect of amorolfine nail lacquer”. Mycoses. 48 (2): 91–4. doi:10.1111/j.1439-0507.2004.01090.x. PMID 15743424.
- ^ Feng X, Xiong X, Ran Y (May 2017). “Efficacy and tolerability of amorolfine 5% nail lacquer in combination with systemic antifungal agents for onychomycosis: A meta-analysis and systematic review”. Dermatologic Therapy. 30 (3): e12457. doi:10.1111/dth.12457. PMID 28097731.
- ^ It can readily be verified that Curanail is advertised on websites such as US Amazon.com, shipped from abroad.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | D01AE16 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78613-35-1 |
| PubChem CID | 54260 |
| ChemSpider | 49010 |
| UNII | AB0BHP2FH0 |
| KEGG | D02923 |
| ChEBI | CHEBI:599440 |
| ChEMBL | ChEMBL489411 |
| CompTox Dashboard (EPA) | DTXSID0046690 |
| Chemical and physical data | |
| Formula | C21H35NO |
| Molar mass | 317.517 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////AMOROLFINE, Ro 14-4767-002, аморолфин ,أمورولفين ,阿莫罗芬 , antifungal
