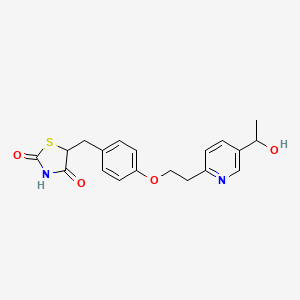
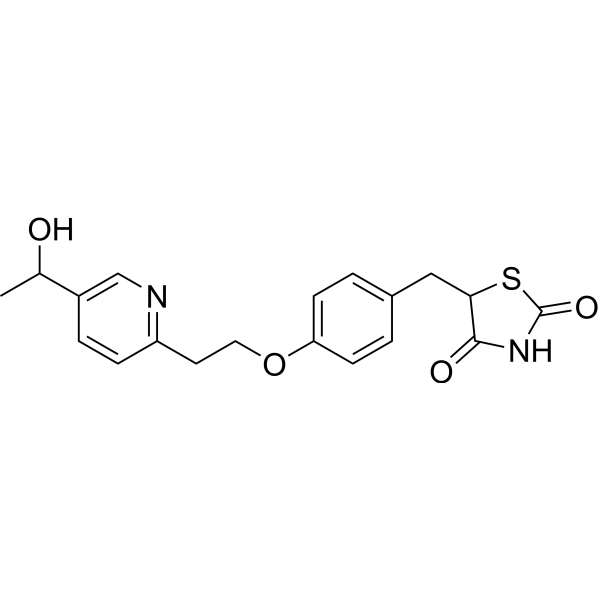
LERIGLITAZONE
C19H20N2O4S,
| MW 372.4 |
Hydroxypioglitazone, CAS 146062-44-4
MIN 102, Hydroxy Pioglitazone (M-IV)лериглитазон [Russian] [INN]ليريغليتازون [Arabic] [INN]乐立格列酮 [Chinese] [INN]
5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione
Hydroxypioglitazone is a member of the class of thiazolidenediones that is the hydroxy derivative of pioglitazone. It has a role as a human xenobiotic metabolite. It is a member of thiazolidinediones, a member of pyridines and an aromatic ether. It derives from a pioglitazone.
- OriginatorIDIBELL
- DeveloperMinoryx Therapeutics
- ClassNeuroprotectants; Phenyl ethers; Pyridines; Small molecules; Thiazolidinediones
- Mechanism of ActionPeroxisome proliferator-activated receptor gamma agonists
- Orphan Drug StatusYes – Adrenoleucodystrophy; Friedreich’s ataxia
- Phase II/IIIAdrenoleucodystrophy
- Phase IIFriedreich’s ataxia
- PreclinicalCNS disorders
- 23 Sep 2020Leriglitazone receives Rare Pediatric Disease designation from the US FDA for X-linked adrenoleukodystrophy before September 2020
- 23 Sep 2020Minoryx Therapeutics licenses leriglitazone to Sperogenix Therapeutics in China, Hong Kong and Macau for X-linked adrenoleukodystrophy (X-ALD)
- 14 Sep 2020Minoryx Therapeutics completes the phase II FRAMES trial in Friedreich’s ataxia (In adolescents, In adults) in Spain, Germany, France and Belgium (PO) (NCT03917225)

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Leriglitazone (Hydroxypioglitazone), a metabolite of pioglitazone. Leriglitazone (Hydroxypioglitazone) PioOH is a PPARγ agonist, stabilizes the PPARγ activation function-2 (AF-2) co-activator binding surface and enhances co-activator binding, affording slightly better transcriptional efficacy. Leriglitazone (Hydroxypioglitazone) binds to the PPARγ C-terminal ligand-binding domain (LBD) with Ki of 1.2 μM,induces transcriptional efficacy of the PPARγ (LBD) with EC50 of 680 nM.
Leriglitazone is under investigation in clinical trial NCT03917225 (A Clinical Study to Evaluate the Effect of MIN-102 on the Progression of Friedreich’s Ataxia in Male and Female Patients).
Treatment of X-Linked Adrenoleukodystrophy
PATENT
WO 9218501
WO 9322445
PAPER
Chemical & Pharmaceutical Bulletin (1995), 43(12), 2168-72
https://www.jstage.jst.go.jp/article/cpb1958/43/12/43_12_2168/_article
The metabolites of (±)-5-[p-[2-(5-ethyl-2-pyridyl)ethoxy]benzyl]-2, 4-thiazolidinedione (1, pioglitazone), which is a representative insulin-sensitizing agent, were synthesized to confirm their structures and for studies of their pharmacological properties. Of the metabolites identified, a compound hydroxylated at the 2-position of the ethoxy chain (3) and compounds oxygenated at the ethyl side chain attached to the pyridine ring (4, 5) were found to be active, although the potency was slightly lower than that of the parent compound.




PAPER
Journal of Medicinal Chemistry (1996), 39(26), 5053-5063.
https://pubs.acs.org/doi/10.1021/jm9605694
Pioglitazone (5-(4-(2-(5-ethyl-2-pyridyl)ethoxy)benzyl)-2,4-thiazolidinedione, 2) is a prototypical antidiabetic thiazolidinedione that had been evaluated for possible clinical development. Metabolites 6−9 have been identified after dosing of rats and dogs. Ketone 10 has not yet been identified as a metabolite but has been added to the list as a putative metabolite by analogy to alcohol 6 and ketone 7. We have developed improved syntheses of pioglitazone (2) metabolites 6−9 and the putative metabolite ketone 10. These entities have been compared in the KKAy mouse model of human type-II diabetes to pioglitazone (2). Ketone 10 has proven to be the most potent of these thiazolidinediones in this in vivo assay. When 6−10 were compared in vitro in the 3T3-L1 cell line to 2, for their ability to augment insulin-stimulated lipogenesis, 10 was again the most potent compound with 6, 7, and 9 roughly equivalent to 2. These data suggest that metabolites 6, 7, and 9 are likely to contribute to the pharmacological activity of pioglitazone (2), as had been previously reported for ciglitazone (1).
PATENT
WO 2015150476
Compound 5-[4-[2-(5-(1 -hydroxyethyl)-2-pyridinyl)ethoxy]benzyl]-2,4-thiazolidinedione of formula (1 ) can be prepared according to Scheme 1 (see e.g. J.Med.Chem. 1996, 39(26),5053).
Scheme 1
Scheme 2
Yet another method to prepare mixtures (c) – comprising compound (2) and (4) – and (d) – comprising compounds (3) and (5) – (scheme 3), includes the resolution of the racemic mixture VIII using the already described methods (chiral HPLC separation, enzymatic resolution, chiral resolution, etc) followed by double bond reduction in each of the enantiomers Villa and Vlllb.
Scheme 4
Compounds of formula (2), (3), (4) and (5) may be obtained from mixtures (c) and (d) (Scheme 45) by chiral HPLC separation. Alternatively, the desired enantiomerically pure compounds can be prepared by chiral synthetic procedures known to those skilled in the art (for example: asymmetric hydrogenolysis of the corresponding single isomer of compound VI).
HPLC Method
Column: Symmetry Shield RP-18, 5 μηη (4.6 x 250 mm); wavelength: 210 nm; flow: 1 mL/min; run time: 28 min; mobile phase-gradient: (t/%B): 0/10, 8/10, 12/60, 16/80, 20/80, 24/10, 28/10 [A: Water (potassium dihydrogen o-phosphate (pH~3)), B: Acetonitrile]
A mixture of compounds (2) and (4) (mixture (c)) and a mixture of compounds (3) and (5) (mixture (d)) were prepared according to Scheme 7.
Example 6: Preparation of diastereomeric mixtures D-1 and D-2 of M-IV:
Scheme 1 :
Ent-1 (VIII) Ent-2 (VIII)
Step 3 Step 3
MIV D-1 MIV D-2
Step 1 : Synthesis of compound VIII: HCI (48 ml, 2N) was added to a solution of compound VI (10 g, 0.024 mol) in methanol (200 ml) and the mixture was heated to reflux. After 4 h of reflux, the reaction mixture was cooled to r.t. and concentrated under reduced pressure to afford a yellow solid. The solid was suspended in water (70 ml) and neutralized using a saturated NaHC03 solution. The resulting pale yellow precipitate was collected by filtration and vacuum dried to afford compound VIII (7.5 g; 84% yield).
ES-MS [M+1]+: 371.0.
Step 2: Chiral prep. HPLC
Compound VIII (1 .0 g) was dissolved in a mixture containing equal volumes of acetonitrile, methanol and dichloromethane; injected (150 μΙ injections) in chiral prep-HPLC column (Chiralpak-IA 250 x 20 mm, 5 micron) and separated [Mobile phase- n-Hexane/0.05% Et3N in EtOH (50:50); flow Rate: 18ml/min; run time: 60 min]. The fractions containing the enantiomers Villa and Vlllb were separately concentrated under reduced pressure to minimum volume and the respective residues were diluted with EtOAc (100 ml), followed by water (50 ml). The resultant organic phases were
dried over anhydrous Na2S04 and concentrated to afford compounds Villa and Vlllb as off-white solids. Enantiomers Villa and Vlllb were isolated but the absolute configuration of each enantiomer has not been determined.
Compound Ent-1 (VIII): 250 mg (Yield: 50%); tR (Chiral HPLC) = 14.8 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 9.2 Hz, 2H), 7.31 (d, J = 7.6 Hz, 1 H), 7.08 (d, J = 8.8 Hz, 2H), 5.25 (d, J = 4.0 Hz, 1 H), 4.74-4.76 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (t, J = 6.4 Hz, 2H), 1.34 (d, J = 6.4 Hz, 3H).
Compound Ent-2 (VIII): 237 mg (Yield: 47%); tR (Chiral HPLC) = 16.7 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 8.8 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1 H), 7.08 (d, J = 9.2 Hz, 2H), 5.23 (d, J = 3.6Hz, 1 H), 4.75 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (dd, J = 6.8, 6.4 Hz, 2H), 1 .34 (d, J = 6.4 Hz, 3H).
Synthesis of diastereomeric mixtures of M-IV
Synthesis of D-1 MIV
Step 3: A solution of NaBH4 (77 mg, 2.02 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-1 (VIII) (250 mg, 0.675 mmol), dimethylglyoxime (32 mg, 0.27 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) in a mixture of water (10 ml), THF (10 ml) and 1 M NaOH (0.5ml) solution at 10 °C, and the reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (26 mg, 0.675 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) were added and stirring was continued at r.t. [additional quantities of CoC|2 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 90-96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-1 (125 mg) as off-white solid.
Synthesis of D-2 MIV
Step 3: A solution of NaBH4 (72 mg, 1 .921 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-2 (VIII) (237 mg, 0.64 mmol), dimethylglyoxime (30 mg, 0.256 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) in a mixture of water (10 ml), THF (10 ml), and 1 M NaOH (0.5ml) solution at 10 °C, and the
reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (24 mg, 0.64 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) were added and stirring was continued at r.t. [additional quantities of CoCI2.6H20 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound, which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-2 (100 mg) as off-white solid.
MIV D-1 : yield: 125 mg (50%); tR (Chiral HPLC) = 17.8, 14.7 min; ES-MS [M+1]+: 373.0, 1H NMR (400 MHz, DMSO-d6): δ 12.00 (br s, NH), 8.46 (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.4 Hz, 1 H), 7.30 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1 .34 (d, J = 6.4 Hz, 3H).
MIV D-2: yield: 100 mg (42%); tR (Chiral HPLC) = 19.4, 16.5 min; ES-MS [M+1]+: 373.0; 1H NMR (400 MHz, DMSO-d6): δ 12.01 (br s, -NH), (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (dd, J = 6.8, 6.4 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1.34 (d, J = 6.8 Hz, 3H).
Diastereomeric mixtures D-1 and D-2 of MIV correspond to mixtures (c) and (d) described above, but the specific diastereomers present in each diastereomeric mixture have not been assigned.
Example 7: in vitro ADME and toxicological characterization
Protocol: The assays performed include cytochrome P450 inhibition with the different isoforms, microsomal and hepatocyte stability, neurotoxicity in neural cells and hERG safety assays using a patch clamp electrophysiology measurement (FDA Draft Guidance for Industry. Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations 2012, The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions Adopted in 2012, Schroeder K et al. 2003 J Biomol Screen 8 (1 ); 50-64, Barter ZE et al. 2007
Curr Drug Metab 8 (1 ); 33-45, LeCluyse EL and Alexandre E 2010 Methods Mol Biol 640; 57-82). The results indicate a safe and favourable ADME profile for the compounds of the invention.
Example 8: The brain plasma ratios of Pioglitazone, MIV, Mill and Mil following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice.
The brain-plasma ratio was calculated based on levels of Pioglitazone, MIV, Mill and Mllin plasma and brain quantified at C max (maximal concentration) following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice. The percentage brain plasma ratio was 9, 13, 7 and 1 %, respectively, for Pioglitazone, Mil and Mill as shown in the Figure 4. Thus, active metabolites Mill and Mil crossed the BBB at much lower extent than Pioglitazone as it was predicted based on the physicochemical properties of the compounds (see Tablel ). In contrast, unexpectedly metabolite MIV crossed the BBB in a higher percentage than the parent compound Piolgitazone
The calculations of the both indexes (ClogP and QPIogBB) for Pioglitazone and its metabolites Mil and Mill are shown in Table 1 . For both indexes the 2 metabolites are lower than for pioglitazone, suggesting for Mil, and Mill a less favored penetration and distribution within CNS.
TABLE 1
PATENT
WO 2018116281
https://patents.google.com/patent/WO2018116281A1/enPioglitazone is a “dirty” drug which is converted to many metabolites in vivo. The metabolic pathway of pioglitazone after oral administration has been studied in several animal species and in humans and the metabolites have been described in the literature (see e.g. Sohda et al, Chem. Pharm. Bull., 1995, 43(12), 2168-2172) and Maeshiba et al, Arzneim.-Forsch/Drug Res, 1997, 47 (I), 29-35). At least six metabolites have been identified, named M-I to M-VI. Amongst these metabolites, M-II, M-III and M-IV show some pharmacological activity but are less active than Pioglitazone in diabetic preclinical models.
[0005] 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione has the following structure:

[0006] Tanis et al. (J. Med. Chem. 39(26 ):5053-5063 (1996)) describe the synthesis of 5-[[4-[2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione as follows:Scheme 1

[0007] Tanis et al. describe that the intermediate 14 was obtained in a 27% yield by reacting compound 13 in an aqueous 37% formaldehyde at 170°C for 6 hours. In this process, 5-[[4- [2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (compound 6 in Scheme 1) was obtained in a 2.47% overall yield.[0008] WO 2015/150476 Al describes the use of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts, in the treatment of central nervous system (CNS) disorders. WO 2015/150476 Al describes that 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione was prepared according to the process of Tanis et al. (supra) where the intermediate corresponding to compound 14 of Tanis et al. was prepared similarly at 160°C for 5 hours providing a 17% yield. The overall yield of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione was about 1.5%.[0009] Due to the low yield of the intermediate 2-[5-(l-methoxymethoxy-ethyl)pyridine-2- yl]ethanol, the process step for preparing this intermediate is critical for the overall yield of the product, 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione. In addition, the prior art process to obtain compound 14 is difficult to scale because the reaction is carried out in a pressure vessel at a very high temperature and it is a very dirty reaction.[0010] Accordingly, the processes described in the art afford the product 5-[[4-[2-[5-(l- hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione only in a very low overall yield and, therefore, they are not suitable for large scale synthesis. In addition, the prior art process employs CH3OCH2CI, a known carcinogen, for protecting the hydroxyl group in the key intermediate. There is a need for an improved process for synthesizing 5- [[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts.Formula I illustrated by Scheme 2:Scheme 2 r
B


deprotectionoptional saltformation

I (HCI salt)[0255] In another embodiment, the disclosure provides a process for preparing the compound of Formula I illustrated by Scheme 3 : Scheme 3C
Br. e

step ‘< step b step c

step step g

[0256] In another embodiment in Scheme 3, step c, the order of mixing of the reagents can be as follows: 1. n-BuLi, 2. ethylene oxide, and 3. Cul. This order of mixing is described in Example 2.[0257] In the step a, 2,5-dibromopyridine (1) is reacted with i-PrMgCl in THF and then further with acetaldehyde to obtain compound 2. The reaction mixture is preferably filtered over Celite® after the reaction to remove most of the salts. In one embodiment, the addition of acetaldehyde is conducted at a temperature between -15°C and -10°C to control the exothermic reaction. [0258] In the step b, compound 2 is reacted with TBDMS-C1 in the presence of imidazole having DMF as a solvent. The crude product 3 is advantageously purified by a short plug filtration.[0259] In the step c, the hydroxyl protected compound 3 is reacted with ethylene oxide in the presence of n-BuLi and Cu(I)iodide while maintaining the reaction temperature, i.e., the reaction mixture temperature, below -20°C. In one embodiment, the reaction temperature is maintained below -55°C while adding n-BuLi and Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi, followed by ethylene oxide and then Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi into the reaction mixture, followed by ethylene oxide. In this embodiment, Cu(I)iodide is added then into the reaction mixture while the reaction mixture temperature is maintained below -20°C, and preferably below -55 °C. The reaction mixture is then allowed to slowly warm to room temperature after the addition of the reagents and stirred at room temperature, e.g., 20-25°C, overnight. This process is described in detail in Example 2. After the reaction, the complexed copper is advantageously removed by washing with 10% ammonia. The crude compound 4 can be purified by column chromatography to give >99% pure product with a yield of about 52%.[0260] The following examples are illustrative, but not limiting, of the methods of the present invention. Suitable modifications and adaptations of the variety of conditions and parameters normally encountered in clinical therapy and which are obvious to those skilled in the art in view of this disclosure are within the spirit and scope of the invention.ExamplesCOMPARATIVE EXAMPLE 1Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione (9a) according to the process described in WO 2015/150476 Al Scheme 4

8a 9a[0261] (a) Synthesis of l-(6-methyl-pyridin-3-yl)-ethanol (3a)[0262] LiHMDS (1.0 M in tetrahydrofuran, 463 ml, 0.463 mol) was added drop wise to a cooled solution of methyl 6-methylnicotinate (la) (20 g, 0.132 mol) and ethyl acetate (82 g, 0.927 mol) in dimethylformamide at -50°C; gradually raised the temperature to room temperature and stirred at the same temperature. After 1 h, the reaction mixture was cooled to 0°C; slowly diluted with 20% sulphuric acid and heated to reflux. After 4 h, the reaction mixture was cooled to room temperature, and further to 0°C and basified with potassium carbonate. The reaction medium was diluted with water and extracted in ethyl acetate (3×50 mL). Combined organic extract was dried over sodium sulphate and concentrated to afford crude l-(6-methylpyridin-3-yl)ethan-l-one (2a) (20.0 g) which was taken to the next step without any purification. ES-MS [M+l]+: 136.1.Sodium borohydride (2.3 g, 0.06 mol) was added in small portions over 30 min, to a solution of compound 2a (16.4 g, 0.121 mol) in ethanol (160 mL) at 0°C and the reaction mixture was stirred at same temperature. After 1 h, the reaction mixture was diluted with sodium bicarbonate solution (sat) (2×200 mL) and extracted with dichloromethane (2×500 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford a pale yellow oil, which was purified by flash column chromatography (5% methanol/dichloromethane) to afford compound 3a (17.0 g; 93% yield over 2 steps) as a pale yellow oil. ES-MS [M+l]+: 138.1. 1H NMR (400 MHz, CDC13): δ 8.35 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.0, 2.4 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 4.89 (q, J = 6.5 Hz, 1H), 3.30 (br s, 1H), 2.50 (s, 3H), 1.48 (d, J = 6.5 Hz, 3H).[0263] (b) Synthesis of 5-(l-methoxymethoxy-ethyl)-2-methyl-pyridine (4a):Compound 3a (15 g, 0.109 mol) was added, drop wise, to a cooled suspension of sodium hydride (6.56 g, 0.164 mol) in tetrahydrofurane (150 mL) and stirred at 0°C. After 30 min, chloromethyl methyl ether (13.2 g, 0.164 mol) was added drop wise while stirring and keeping the internal temperature around 0°C. After addition is over, the reaction mixture was stirred at the same temperature for 1 h. The reaction was quenched with ice cold water (80 mL) and extracted with ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford an orange color oil, which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 4a (10.0 g; 51% yield) as a pale yellow oil. ES-MS [M+l]+: 182.2. 1H NMR (400 MHz, CDC13): δ 8.45 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.0, 2.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 4.75 (q, J = 6.4 Hz, 1H), 4.57 (ABq, 2H), 3.36 (s, 3H), 2.53 (s, 3H), 1.48 (d, J = 6.6 Hz, 3H).[0264] (c) Synthesis of 2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethanol (5a):A mixture of compound 4a (7.0 g, 0.0386 mol) and 37% formaldehyde solution (5.8 g, 0.077 mol) was heated to 160°C in a sealed glass tube for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford a crude compound which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 5 (1.2 g; 17% yield) as pale yellow oil. ES-MS [M+l]+: 212.1. 1H NMR (400 MHz, CDC13): δ 8.42 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 8.0, 2.4 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.72 (q, J = 6.6 Hz, 1H), 4.65 (t, J = 5.6 Hz, 1H), 4.52 (ABq, 2H), 3.73 (m, 2H), 3.24 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 1.49 (d, J = 6.4 Hz, 3H).[0265] The total yield for compound 5a from compound la was 8% molar.[0266] (d) Synthesis of 4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzaldehyde (6a): Methanesulphonylchloride (1.19 g, 0.01 mol) was added, drop wise, to a cooled suspension of compound 5a (1.7 g, 0.008 mol) and triethylamine (1.79 ml, 0.013 mol) in dichloromefhane (20 mL) at 0°C and stirred at same temperature for 1 h. The reaction mixture was diluted with water (50 mL) and extracted with dichloromethane (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate (2.04 g; 88% yield) as a yellow oil, which was taken to next step without purification. ES-MS [M+l]+: 290.[0267] 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate was added (2.3 g, 0.008 mol) to a stirred suspension of 4-hydroxybenzaldehyde (1.65 g, 0.0137 mol) and potassium carbonate (1.86 g, 0.0137 mol) in mixture of toluene (25 mL) and ethanol (25 mL); stirred at 85°C for 5 h. After consumption of the starting materials, the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate (2×100 mL). The combined organic extract was washed with water; dried over anhydrous sodium sulphate and concentrated to afford a crude dark yellow liquid. The crude was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 6a (1.5 g; 60% yield) as pale yellow liquid. ES-MS [M+l]+: 316.1.[0268] (e) Synthesis of 5-(4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzylidene)-thiazolidine-2,4-dione (7a):Piperidine (80 mg, 0.95 mmol) was added to a solution of compound 6a (0.6 g, 1.9 mmol) and thiazolidine-2,4-dione (0.22 g, 1.9 mmol) in ethanol (15 mL) and the mixture was heated to reflux overnight. After 15 h, the reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford crude mixture, which was purified by flash column chromatography (2% methanol/dichloromethane) to afford compound 7 (500 mg; 64% yield) as a yellow solid. ES-MS [M+l]+: 415.1. 1H NMR (400 MHz, DMSO-d6): δ 12.25 (br s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 7.70 (dd, J = 8.0, 2.0 Hz, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 4.73 (m, 1H), 4.60-4.40 (m, 4H), 4.22 (t, J = 6.2 Hz, 1H), 3.24 (s, 3H), 3.20 (t, J = 6.8 Hz, 2H), 1.41 (d, J = 6.0 Hz, 3H).[0269] (f) Synthesis of 5-(4-{2-[5-(l-hydroxy-ethyl)-pyridin-2-yl]-ethoxy}-benzyl)- thiazolidine-2,4-dione (9a): [0270] A solution of sodium borohydride (115 mg, 3.017 mmol) in 0.2N sodium hydroxide(1.2 mL) was added slowly to a stirred solution of compound 7 (0.5 g, 1.207 mmol), dimethylglyoxime (42 mg, 0.36 mmol) and C0CI2.6H2O (23 mg, 0.096 mmol) in a mixture of water (6 mL): tetrahydrofurane (6 mL) and 1M sodium hydroxide (1 mL) solution at 10°C and after addition, the reaction mixture was stirred at room temperature. After 1 h, the reaction color lightened and additional quantities of sodium borohydride (46 mg, 1.207 mmol) and C0CI2.6H2O (22 mg, 0.096 mmol) were added and stirring was continued at room temperature. After 12 h, the reaction was neutralized with acetic acid (pH~7); diluted with water (10 mL) and extracted in ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford crude compound 8a, 5-(4- (2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethoxy)benzyl)thiazolidine-2,4-dione, (0.4 g) as pale yellow semi solid, which was taken to next step without purification. ES-MS [M+l]+: 417.5.[0271] 2N HC1 (2 mL) was added to a solution of compound 8a (0.4 g, 0.96 mmol) in methanol (20 ml) and the mixture was heated to reflux. After 4 h, the reaction mixture was cooled to room temperature and then concentrated under reduced pressure to afford a residue which was dissolved in water and the solution was neutralized using sodium bicarbonate solution (sat). The resulting white precipitate was collected by filtration to afford compound 9a (250 mg; 56% yield over 2 steps) as an off-white solid. ES-MS [M+l]+: 373.4. 1H NMR (400 MHz, DMSO-de): δ 12.00 (br s, -NH), 8.46 (d, J = 2.0 Hz, 1H), 7.66 (dd, J = 8.0, 2.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.25 (d, J = 4.4 Hz, 1H), 4.86 (m, 1H), 4.75 (m, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1H), 3.14 (t, J = 6.4 Hz, 2H), 3.04 (m, 1H), 1.34 (d, J = 6.4 Hz, 3H).[0272] The overall yield of compound 9a was 1.5% molar.EXAMPLE 2Synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol[0273] The synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol was conducted according to the Scheme 5 using the reagents and solvents listed in Table 1 below: Scheme 5TBDMS-CI OTBDMS 1 . n-BuLi, <-55°C OTBDMSImidazole
DMF

[0274] The 1H-NMR spectra were recorded with Agilent MercuryPlus 300 NMR spectrometer.[0275] LC-MS data were obtained on an Agilent 1290 series with UV detector and HP 6130MSD mass detector using as column Waters XB ridge BEH XP (2.1 x 50 mm; 2.5 μιτι) and as eluent Ammonium acetate (10 mM); Water/ Methanol/ Acetonitrile.[0276] (a) l-(6-bromopyridin-3-yl)ethan-l-ol (2)[0277] A 20 L vessel was placed under nitrogen atmosphere and charged with tetrahydrofuran (5.5 L) and 2,5-dibromopyridine (1) (2000 g, 8.44 mol, 1.0 eq) (OxChem Corporation). The mixture was cooled to -10°C and isopropyl magnesium chloride (20% in THF, 6.02 L, 11.82 mol, 1.4 eq) (Rockwood Lithium) was added slowly over 1 h, keeping the reaction temperature below 5°C. After addition, the cooling bath was removed and the temperature was kept below 30°C (some additional cooling was needed to achieve this) and the reaction mixture was stirred overnight. After 16 h, a sample was taken; quenched with saturated aqueous ammonium chloride and extracted with methyl tert-buty\ ether (TBME). The TBME was evaporated under vacuum. 1H-NMR in deuterated chloroform showed complete conversion.[0278] The reaction mixture was cooled to -15°C and a solution of acetaldehyde (472 g,10.72 mol, 1.27 eq) (Acros) in tetrahydrofuran (200 mL) was added dropwise, while keeping temperature below -10°C. After the addition was complete, the cooling bath was removed and the temperature was allowed to rise to maximum of 5-8°C. After 1.5 h, a sample was taken and the reaction was quenched with aqueous ammonium chloride as described above. 1H-NMR showed the reaction was complete.[0279] Two batches were combined for work up.[0280] The reaction mixture was quenched by pouring the mixture into a solution of aqueous ammonium chloride (1 kg in 5 L water) and stirred for 15 min, filtered over Celite and rinsed thoroughly with toluene. The filtrate was transferred to a separation funnel and the obtained two layers system was separated. The aqueous layer was extracted with toluene (2 L). The combined organic layers were dried over sodium sulfate and filtered. Evaporation of the filtrate to dryness under vacuum yielded 3.49 kg (99%) of the desired crude material. XH NMR (300 MHz, CDC13): δ 8.30 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.0, 2.5 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 4,91 (q, J = 6.5 Hz, 1H), 1.49 (d, J = 6.5 Hz, 3H).[0281] (b) 2-bromo-5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridine (3)[0282] A 50 L reactor under nitrogen atmosphere was charged with compound 2 (10.0 kg, around 49.5 mol) and DMF (16 L). The mixture was cooled to 10°C and imidazole (6.74 kg, 99 mol, 2.0 eq) (Apollo Scientific Ltd.) was added portion wise within 30 min. The mixture was cooled to 0°C and TBDMS-Cl (7.46 kg, 49.5 mol, 1.0 eq) (Fluorochem) was added portion wise within 5 h, keeping the temperature below 3°C. The mixture reaction temperature was allowed to reach room temperature and stirred overnig ht. H NMR of a sample showed complete conversion.[0283] The reaction mixture was transferred to a 100 L extraction-vessel and the product was extracted with heptane (2×7.5 L, 10 L). The combined heptane-layers were washed with water (2×6 L, 3 L) to remove small amounts of DMF, dried over sodium sulfate and evaporated under vacuum to give crude compound 3 (15.5 kg, 49.0 mol) in a 99.0% yield. This crude product was purified by a short plug filtration, using 10 kg silica/heptane and eluted with heptane (approx. 50 L). The product-fractions were combined and evaporated under vacuum to give 12.0 kg of purified compound 3 (38 mol) as a brown oil in a 76.8% molar yield. (Average yield for 3 experiments was 78%). HPLC-MS: Rt= 2.6 min, M+l=316.1 and 318.1; 1H NMR (300 MHz, CDC13): δ 8.55 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 8.2, 2.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 4,86 (q, J = 6.5 Hz, 1H), 1.40 (d, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0284] (c) 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol (4)[0285] The ethylene oxide solution in diethylether was prepared in advance. Diethylether(1.2 L) in a 3 L three-necked flask was cooled at -65 °C and ethylene oxide (462.3 g, 10.5 mol, 1.06 eq) (Linde) was added and stirred at -70°C. Alternatively, the ethylene oxide solution can be made at about -20°C and then added gradually to the reaction mixture having a temperature at about -60°C. [0286] To a solution of 2-bromo-5-(l-((ieri-butyldimethylsilyl)oxy)ethyl)pyridine (3) (3.13 kg, 9.90 mol, 1.0 eq) in diethylether (7.5 L) cooled at -59°C, n-butyllithium (4 L, 10.0 mol, 2.5M in hexanes, 1.01 eq) (Aldrich Chemistry) was added while keeping temperature between -58°C and -62°C. After addition, the mixture was stirred for 1 h while keeping temperature between -60°C and -68°C. The upfront prepared ethylene oxide solution was added at once to the reaction mixture, while temperature was around -62°C. Subsequently, copper(I) iodide (962.3 g, 5.05 mol, 0.51 eq) (Acros Organics) was added in portions of 120 g, every 10 min, keeping the temperature between -61°C and -63°C. Stirring was continued for 1 h after addition keeping temperature between -61°C and -63°C. The cooling bath was removed and allowing the temperature to rise to about 15°C and further to 25 °C with a water bath overnight.[0287] Workup: The reaction-mixture was poured into a solution of 1 kg ammonium- chloride in 5 L water and stirred for 30 min, then the layers were separated. The organic layer was washed with aqueous ammonium hydroxide (10%, 2.5 L, 4x) to remove Cu-complex (blue color disappeared). The combined organic layers were dried over sodium sulfate and evaporated to give 3.12 kg (max. 9.90 mol) crude compound 4 as a brown oil. The crude compound was purified over 20 kg silica (heptane/EtOAc) by eluting with 80 L heptane/EtOAc, 20 L EtOAc, 25 L EtOAc/MeOH 95/5, 25 L EtOAc/MeOH 9/1 and 10 L EtOAc/MeOH 8/2, to give 1.47 kg of purified compound 4 (5.22 mol) as a brown oil (with tendency to solidify) in a 52.7% average molar yield (HPLC-purity of 99.5%). (Average yield over 12 experiments 52%). HPLC-MS: Rt= 2.3 min, M+l=282.1; 1H NMR (300 MHz, CDC13): δ 8.42 (d, J = 2.1 Hz, 1H), 7.61 (dd, J = 8.3, 2.1 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 4,88 (q, J = 7.0 Hz, 1H), 4.01 (t, J=6.0 Hz, 2 H), 3.00 (t, J=6.0 Hz, 2 H), 1.41 (d, J =7.0 Hz, 3H), 0.90 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0288] Another 2.5% of the product was isolated by re -purifying impure product fraction.The total yield of compound 4 from compound 1 was 39.6% molar.EXAMPLE 3Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione hydrochloride (9) 2. Sodium bisulfiteethanol/water mixture
3. Addition 10% aqueous sodium hydroxide solution

until pH 12

from step e dimethylglyoxime7step g step f

step h[0289] The 1H-NMR spectra were recorded with a 400 MHz Avance Bruker NMR spectrometer. LC-MS data were obtained on a Agilent Technologies 6130 Quadrapole LC/MS using as column Agilent XDB-C18 and as eluent 0.1% formic acid (aq) and 0.05% formic acid in acetonitrile.[0290] Steps d and e: Synthesis of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl] ethoxy] -benzaldehyde (6)[0291] To a well stirred solution of 5-[[[(l,l-dimethylethyl)dimethylsilyl]-oxy]ethyl]-2- pyridineethanol (4) (obtained as described in Example 2) (1.91 kg) in toluene (8.6 L) at 5°C were added sodium hydroxide (30% aqueous, 2.79 L) and tetrabutylammonium bromide (7.2 g). p-Toluenesulfonyl chloride (1.62 kg) was next added in portions during 5 min. After the addition, the reaction mixture was allowed to reach room temperature in 0.5 h and stirred at this temperature for 18 h. Water (7.3 L) was then added and the mixture was mixed well. Once the solids were dissolved, the layers were allowed to settle and the organic layer was separated. This organic phase was washed with water (5.7 L, 2x), followed by washing with a solution of sodium chloride (57 g) in water (5.7 L). The solvents were concentrated at reduced pressure to an amount of 2.5 kg of a brown oil (compound 5).[0292] To this well stirred brown oil were added subsequently ethanol (7.8 L), water (0.86L), 4-hydroxybenzaldehyde (0.88 kg) and potassium carbonate (1.17 kg) and then the mixture was heated at 75 °C for 18 h. Then, the solvent was evaporated while adding toluene (7.7 L) during 6 h and then the reaction mixture was allowed to cool. At 30°C, water (7.6 L) was added, stirred until all solids were dissolved and the mixture was cooled to room temperature. The layers were allowed to settle and separated. The organic layer was washed with water (7.6 L). The first aqueous extract was extracted with toluene (2.8 L) and this organic extract was used to also extract the aqueous washing. The organic extracts were combined and concentrated under vacuum to give 3.49 kg of a black oil (crude title compound 6).[0293] 1.73 kg of this black oil was dissolved in ethanol (0.74 L) and added to a well stirred solution of sodium bisulfite (1.36 kg) in a mixture of water (3.27 L) and ethanol (0.74 L). The container of the black oil was rinsed with ethanol (0.37 L) twice and these two rinses were also added to the bisulfite reaction mixture. After 75 min, heptane (5.3 L) was added, well mixed for 5 min, and the layers were allowed to settle and separated. To the organic layer was added a solution of sodium bisulfite (0.55 kg) in water (2.65 L), and ethanol (1.06 L). After stirring for 30 min, the layers were allowed to settle and separated. The two bisulfide aqueous extracts were combined and flasks rinsed with water (2.12 L). Next, toluene (4.5 L) and heptane (4.5 L) were added, the mixture was well stirred and the pH was adjusted to 12 using sodium hydroxide (10% aq) (temperature became 32°C). After stirring for an additional 5 min, the layers were allowed to settle and separated at 30°C. The aqueous layer was extracted with a mixture of toluene (1.5 L) and heptane (3.0 L). The layers were separated and the organic layers were combined. The combined organic layers were washed with water (5 L, 2x) and concentrated under vacuum to give the purified title compound 6. This procedure was repeated with another 1.73 kg of the black oil (crude title compound 6) to give in total 2.77 kg of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]-benzaldehyde (6) as brown oil which contained 24% m/m of toluene according to 1H NMR (yield = 80%, calculated from compound 4 and corrected for residual toluene). [0294] 1H NMR (CDC13) δ: 0.00 (s, 3H), 0.09 (s, 3H), 0.91 (s, 9H), 1.44 (d, = 6 Hz, 3H),3.30 (t, = 7 Hz, 2H), 4.47 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.99 – 7.30 (m, 3H), 7.62- 7.67 (m, 1H), 7.80 – 7.85 (m, 2H), 8.5- 8.54 (m, 1H) and 9.88 (s, 1H).[0295] LC-MS; rt 7.5 min: ES: M+ 387, 386.[0296] Step f: Synthesis of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7)[0297] A solution of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]- ethoxy]-benzaldehyde (6) (2.75 kg, containing 24% m/m of toluene) and piperidine (6.0 g) in methanol (3.16 L) was concentrated at 40°C under reduced pressure. The residue was dissolved in methanol (10.4 L) and 2,4-thiazolidinedione (759 g) and piperidine (230 g) were added. The mixture was heated at 47°C. After 25 h, the reaction mixture was allowed to cool to room temperature. The mixture was kept at pH 5-6 by adjusting it with acetic acid, if necessary. After a night at room temperature, water (1.56 L) was added and the suspension was stirred at room temperature for additional 2 h. The solids were isolated by filtration, washed with methanol (1 L, 2x) and dried under vacuum to give crude compound 7 (1.65 kg). The crude compound was mixed with methanol (10 L) and dichloromethane (8.6 L) and heated at 32°C until all solids dissolved. Then, the solvents were removed by distillation until the temperature of the mixture reached 34°C at a pressure of 333 mbar. Then, it was allowed to cool to room temperature overnight and stirred at 2°C for additional 2 h. The solids were isolated by filtration, washed with methanol (0.5 L, 2x) and dried under vacuum to give title compound 7 (1.50 kg) (yield = 61%).[0298] 1H NMR (CDCI3) δ 0.00 (s, 3H), 0.08 (s, 3H), 0.90 (s, 9H), 1.43 (d, = 6 Hz, 3H),3.32 (t, = 7 Hz, 2H), 4.48 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.95 – 7.00 (m, 2H), 7.24 – 7,28 (m, 1H), 7.38 – 7.42 (m, 2H), 7.67 (s, 1H), 7.69 – 7.73 (m, 1H) and 8.48 (d, = 3 Hz, 1H).[0299] LC-MS; rt 7.5 min: ES: M+ 487, 486, 485.[0300] Step g: Synthesis of 5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (8)[0301] To a stirred suspension of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]- ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7) (10 g) in THF (10 mL) and sodium hydroxide (IN aq, 21 mL) was added of a solution of cobalt chloride (26 mg) and of dimethylglyoxime (930 mg) in THF (2.3 mL) and water (1.0 mL). Then the suspension was put under a nitrogen atmosphere by applying the sequence of vacuum and flushing with nitrogen (4x). Thereafter, the suspension was heated to 30°C. Then, a stock solution of sodium borohydride was prepared by dissolving sodium borohydride (2.7 g) in a mixture of water (15.8 mL) and a solution of sodium hydroxide (1 N aq, 3.5 mL), which was put under a nitrogen atmosphere by applying a sequence of vacuum and flushing with nitrogen (3x). This was added to the suspension of compound 7 at a rate of 4.5 mL/h. Simultaneously, nitrogen gas-saturated acetic acid was added to the suspension at a rate of 0.7 mL/h to maintain a pH of 10.0-10.5. After 1 h 30 min the rate of addition of the sodium borohydride solution and acetic acid were both reduced by half. Next, 3 h 45 min after start of addition, the addition of sodium borohydride and acetic acid were stopped. The mixture was allowed to cool down to room temperature and acetone (2.5 mL) was added over a period of 1 minute. After stirring the reaction mixture for 15 min acetic acid was added until the pH was 5.5-6.0 (about 3 mL required). Next, a mixture of ethyl acetate/toluene (1/3 v/v, 30 mL) was added, well mixed and layers were allowed to settle. The aqueous layer was separated and washed with ethyl acetate/toluene (1/3 v/v, 10 mL). Both organic extracts were pooled and water (40 mL) was added, well mixed and layers were allowed to settle. The pH of the aqueous layer was adjusted to 5.5-6 using saturated sodium hydrogen carbonate solution (aq) and again mixed with the organic layer. Layers were allowed to settle and the organic layer was separated and concentrated under vacuum to give 11.09 g of yellow oil (crude mixture containing title compound 8 and its borane complex). Several batches were combined for work up.33.1 g of the crude mixture containing title compound 8 and its borane complex (not corrected for residual solvents) was dissolved in toluene (30 mL) and filtered. The filtrate was submitted to column chromatography (silica gel, gradient of toluene to toluene/ethyl acetate 1/1) to give 30.0 g of mixture of 5-[[4-[2-[5-[[[(l,l- dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione (8) and its borane complex as a slightly yellow oil (yield = 100% from compound 4, not corrected for residual solvents). [0303] 1H NMR (CDC13) δ: -0.03 – 0.10 (m, 6H), 0.87 – 0.93 (m, 9H), 1.42 (d, / = 6 Hz, 3H),3.05-3.71 (m, 4H), 4.30 – 4.51 (m, 3H), 4.87 – 4.94 (m, 1H), 6.82 – 6.88 (m, 2H), 7.10-7.92 (m, 5H), 8.49 (d, / = 3 Hz, 0.6H) and 8.72 (brs, 0.4H).[0304] LC-MS; rt 6.8 min: ES: M+ 489, 488, 487, M“ 487, 486, 485; rt 8.1 min: ES M“ 501,500, 499, 498, 485.[0305] Step h: Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (9)[0306] To a stirred solution of the mixture of (5-[[4-[2-[5-[[[(l,l-dimethylethyl)- dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and its borane complex (8) (5.17 g) in methanol (25.2 mL) at 22°C was added hydrochloric acid (30%, 2.75 mL) in about 5 min to give a temperature rise to 28°C. This solution was heated to 40 °C. Three hours after addition, the 11 g of volatiles were removed under reduced pressure. Then, acetonitrile (40.3 mL) was added and the mixture was heated at reflux for 0.5 h. Next, the suspension was allowed to cool down to room temperature and stirred for 1 h at room temperature. Solids were isolated by filtration, washed with a mixture of acetonitrile/water (20/1 v/v, 10 mL) and with acetonitrile (10 mL) and dried under vacuum at 40 °C to give 4.00 g of white solids (crude 9) (yield = 77%, not corrected for residual solvents).[0307] Purification of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione hydrochloride (9):[0308] The crude mixture of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (3.95 g, crude 9) was dissolved in methanol/water (7/2 v/v, 80 mL) by heating it to 49°C. To this solution was added washed norit (obtained by heating a suspension of norit (6 g) in methanol/water (7/2 v/v, 90 mL) at 45°C for 1 h, then isolating the norit by filtration and washing it twice with methanol/water (7/2 v/v, 30 mL) and drying it under vacuum at 40°C). Equipment was rinsed with methanol/water (7/2 v/v, 18 mL). After 0.5 h of stirring at 46°C, the warm suspension was filtered to remove the norit and filter was washed twice with methanol/water (7/2 v/v, 18 mL). The filtrate was concentrated under vacuum at a bath temperature of 60°C to a mass of 11.8 g (1 v of compound and 2 v of water). To the suspension was added butanone (19.7 mL, 5 v) and the mixture was heated at a bath temperature of 95°C. Under distillation at a constant volume, butanone (95 mL) was added. Next, heating was stopped and the suspension was allowed to reach room temperature in about 0.5 h. Subsequently it was stirred for 0.75 h at room temperature. The solids were isolated by filtration, washed with a mixture of butanone/water (95/5 v/v, 18 mL) and butanone (18 mL) and dried under vacuum at 40°C to give 3.57 g of compound 9 as white solids (yield = 91%).[0309] 1H NMR (DMSO-de): δ 12.00 (br s, -NH), 8.71 (d, = 2.0 Hz, 1H), 8.45 (dd, = 8.3,1.7 Hz, 1H), 7.98 (d, = 8.3 Hz, 1H), 7.15 (d, = 8.7 Hz, 2H), 6.88 (d, = 8.7 Hz, 2H), 5.57 (s, OH), 4.95 (q, = 6.5 Hz, 1H), 4.86 (dd, = 8.9, 4.4 Hz, 1H), 4.40 (t, = 6.3 Hz, 2H), 3.49 (t, = 6.2 Hz, 2H), 3.29 (dd, = 14.2, 4.4 Hz, 1H), 3.06 (dd, = 14.2, 9.0 Hz, 1H), 1.41 (d, = 6.5 Hz, 3H).[0310] LC-MS; rt 3.5 min: ES: M+ 374, 373, M“ 372, 371.EXAMPLE 4Conditions tested in the preparation of compound 5 in the Step d[0311] The conditions described in Table 2 below were tested in the step d in the preparation of compound 5 from compound 4 providing a good yield of compound 5:Table 2Entry Reaction Conditions Amount of p-Ts-Cl / Eq1 Toluene/water/Bu4NBr/NaOH 1.052 1.083 1.074 1.07+0.035 1.076 Et3N / DCM 1.187 1.408 Pyridine / DCM 1.40 EXAMPLE 5Conditions tested in the preparation of compound 6 in the Step e[0312] The conditions described in Table 3 below were tested in the step e in the preparation of compound 6 from compound 5 providing a good yield of compound 6:Table 3

PATENT
Compound 1 is administered to the subject. The structure of 5-[[4-[2-[5-(l -hydroxy ethyljpyri din-2 – yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione is:
[0047] The present disclosure encompasses the use of stereoisomers of 5-[[4-[2-[5-(l- hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione. 5-[[4-[2-[5- (l-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione has two asymmetric centers and thus four stereoisomers are possible as follows:
//////////LERIGLITAZONE, MIN 102 , лериглитазон , ليريغليتازون , 乐立格列酮 , Hydroxy Pioglitazone, M-IV, PHASE 2
CC(C1=CN=C(C=C1)CCOC2=CC=C(C=C2)CC3C(=O)NC(=O)S3)O
