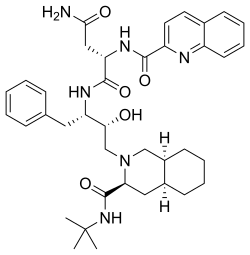
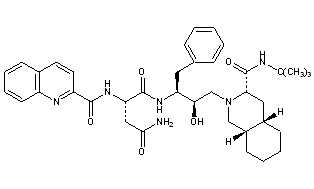
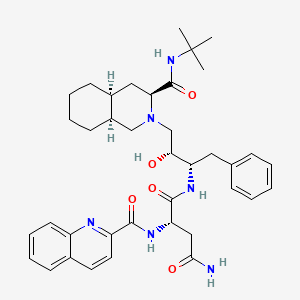
Saquinavir,
Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-decahydroisoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-[(quinolin-2-yl)formamido]butanediamide
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-(quinoline-2-carbonylamino)butanediamide
(-)-cis-N-tert-butyldecahydro-2-{(2R,3S)-2-hydroxy-4-phenyl-3-{[N-(2-quinolylcarbonyl)-L-asparaginyl]amino}butyl}-(3S,4aS,8aS)-isoquinoline-3 carboxamide monomethanesulfonate
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Saquinavir mesylate | UHB9Z3841A | 149845-06-7 | IRHXGOXEBNJUSN-YOXDLBRISA-N |
CAS Registry Number: 127779-20-8
CAS Name: (2S)-N1[(1S,2R)-3-[(3S,4aS,8aS)-3-[[(1,1-Dimethylethyl)amino]carbonyl]octahydro-2(1H)-isoquinolinyl]-2-hydroxy-1-(phenylmethyl)propyl]-2-[(2-quinolinylcarbonyl)amino]butanediamide
Additional Names: (S)-N-[(aS)-a-[(1R)-2-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)octahydro-2(1H)-isoquinolyl]-1-hydroxyethyl]phenethyl]-2-quinaldamido succinamide; N-tert-butyldecahydro-2-[2(R)-hydroxy-4-phenyl-3(S)-[[N-(2-quinolylcarbonyl)-L-asparaginyl]amino]butyl](4aS,8aS)-isoquinoline-3(S)-carboxamide
Manufacturers’ Codes: Ro-31-8959Molecular Formula: C38H50N6O5Molecular Weight: 670.84Percent Composition: C 68.04%, H 7.51%, N 12.53%, O 11.92%
Literature References: Selective HIV protease inhibitor.Prepn: J. A. Martin, S. Redshaw, EP432695; eidem,US5196438 (1991, 1993 both to Hoffmann-LaRoche); K. E. B. Parkes et al.,J. Org. Chem.59, 3656 (1994).In vitro HIV proteinase inhibition: N. A. Roberts et al.,Science248, 358 (1990). Antiviral properties: J. C. Craig et al.,Antiviral Res.16, 295 (1991); S. Galpin et al.,Antiviral Chem. Chemother.5, 43-45 (1994).Clinical evaluation of tolerability and activity: V. S. Kitchen et al.,Lancet345, 952 (1995). Review of pharmacology and clinical experience: S. Kravcik, Expert Opin. Pharmacother.2 303-315 (2001).
Properties: White crystalline solid. [a]D20 -55.9° (c = 0.5 in methanol). Soly (21°): 0.22 g/100 ml water.Optical Rotation: [a]D20 -55.9° (c = 0.5 in methanol)
Derivative Type: Methanesulfonate saltCAS Registry Number: 149845-06-7Additional Names: Saquinavir mesylateManufacturers’ Codes: Ro-31-8959/003Trademarks: Fortovase (Roche); Invirase (Roche)Molecular Formula: C38H50N6O5.CH3SO3HMolecular Weight: 766.95Percent Composition: C 61.08%, H 7.10%, N 10.96%, O 16.69%, S 4.18%
Therap-Cat: Antiviral.Keywords: Antiviral; Peptidomimetics; HIV Protease Inhibitor.
Saquinavir mesylate was first approved by the U.S. Food and Drug Administration (FDA) on Dec 6, 1995, then approved by European Medicine Agency (EMA) on Oct 4, 1996, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Sep 5, 1997. It was developed by Roche, then marketed as Invirase® by Roche in the US and EU and by Chugai in JP.
Saquinavir mesylate is an inhibitor of HIV-1 protease. It is a peptide-like substrate analogue that binds to the protease active site and inhibits the activity of HIV-1 protease that required for the proteolytic cleavage of viral polyprotein precursors into individual functional proteins found in HIV-1 particles. It is indicated for the treatment of HIV-1 infection in combination with ritonavir and other antiretroviral agents in adults (over the age of 16 years).
Invirase® is available as capsule for oral use, containing 200 mg of free Saquinavir. The recommended dose is 1000 mg twice daily in combination with ritonavir 100 mg twice daily for adults.
Human medicines European public assessment report (EPAR): Invirase, saquinavir, HIV Infections, 03/10/1996, 47, Authorised (updated)
EU 08/09/2021
Invirase is an antiviral medicine used to treat adults infected with the human immunodeficiency virus type 1 (HIV 1), a virus that causes acquired immune deficiency syndrome (AIDS). Invirase should only be used in combination with ritonavir (another antiviral medicine) and other antiviral medicines.
Invirase contains the active substance saquinavir.
| Product details | |
|---|---|
| Name | Invirase |
| Agency product number | EMEA/H/C/000113 |
| Active substance | saquinavir |
| International non-proprietary name (INN) or common name | saquinavir |
| Therapeutic area (MeSH) | HIV Infections |
| Anatomical therapeutic chemical (ATC) code | J05AE01 |
| Publication details | |
|---|---|
| Marketing-authorisation holder | Roche Registration GmbH |
| Date of issue of marketing authorisation valid throughout the European Union | 03/10/1996 |
Invirase can only be obtained with a prescription and treatment should be started by a doctor who has experience in the treatment of HIV infection.
Invirase is available as capsules (200 mg) and tablets (500 mg). For patients already taking HIV medicines, the recommended dose of Invirase is 1,000 mg with 100 mg ritonavir twice daily. For patients who are not taking HIV medicines, Invirase is started at 500 mg twice daily with ritonavir 100 mg twice daily for the first 7 days of treatment, given in combination with other HIV medicines. After 7 days, the recommended dose of Invirase is 1,000 mg twice daily with ritonavir 100 mg twice daily in combination with other HIV medicines.
For more information about using Invirase, see the package leaflet or contact a doctor or pharmacist.
The active substance in Invirase, saquinavir, is a ‘protease inhibitor’. It blocks protease, an enzyme involved in the reproduction of HIV. When the enzyme is blocked, the virus does not reproduce normally, slowing down the spread of infection. Ritonavir is another protease inhibitor that is used as a ‘booster’. It slows the breakdown of saquinavir, increasing the levels of saquinavir in the blood. This allows effective treatment while avoiding a higher dose of saquinavir. Invirase, taken in combination with other HIV medicines, reduces the viral load (the amount of HIV in the blood) and keeps it at a low level. Invirase does not cure HIV infection or AIDS, but it may hold off the damage to the immune system and the development of infections and diseases associated with AIDS.
Invirase received a marketing authorisation valid throughout the EU on 4 October 1996.
Drug Name:Saquinavir MesylateResearch Code:Ro-31-8959; Sch-52852Trade Name:Invirase®MOA:HIV-1 protease inhibitorIndication:HIV infectionStatus:ApprovedCompany:Roche (Originator) , ChugaiSales:ATC Code:J05AE01
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2004-12-17 | New dosage form | Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | Priority |
| 1995-12-06 | First approval | Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-10-04 | First approval | Invirase | HIV infection | Capsule | 200 mg | Roche | |
| 1996-10-04 | First approval | Invirase | HIV infection | Tablet, Film coated | 500 mg | Roche |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-09-01 | New dosage form | Invirase | HIV infection | Tablet | 500 mg | Chugai | |
| 1997-09-05 | First approval | Invirase | HIV infection | Capsule | 200 mg | Chugai |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-13 | Marketing approval | 因服雷/Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | |
| 2009-07-01 | Marketing approval | 因服雷/Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche |
Reference:1. US5196438A.Route 2
Reference:1. J. Org. Chem. 1994, 59, 3656-3664.Route 3
Reference:1. WO2006134612A1.
SYN
English: DOI: 10.1021/jo00393a034
DOI: 10.1021/jo00092a026
DOI: 10.1016/S0040-4039(00)77633-7

SYN
In the following, a possible route for the synthesis of Saquinavir is presented. Since Diazomethane is used, the synthesis is not suitable for a scaled up process. Roche has solved this problem with another reaction mechanism. The mechanism for laboratories starts with a ring opening substitution of an epoxid derivative of Phenylalanine with decaisohydroquinoline in dry iso-propanol with nitrogen atmosphere. The intermediate is purified by flash chromatography. In the second step of synthesis, the protection group is removed with gaseous hydrogen and a carbon/palladium catalyst. Furthermore, the new product reacts with N-Benzyloxycarbonyl-Lasparagine(Cbz AsnOH) in the solvents Cbz Asparagine L(Cbz Asn L) and 1- Hydroxybenzotriazolehydrat(HBOT). Afterwards, the protecting group of the former Asparagine is removed with another mixture of gaseous hydrogen and carbon/palladium catalyst. The final intermediate gets stirred in the last step of synthesis together with the solvents Tetrahydrofuran, HBOT and DCC. The mechanism formulated in detail can be found in the Appendix (VIII).7
Kevin E. B. Parkes; David J. Bushnell; et al. Studies toward the Large-Scale Synthesis of the HIV Proteinase Inhibitor Ro 31-8959. J. Org. Chem. 1994, 59, 3656–3664.

SYN
he synthesis of Ro-31-8959/003 (X) was carried out as follows: Condensation of L-phenylalanine (I) with formaldehyde in concentrated hydrochloric acid gave the tetrahydroisoquinoline (II), which was hydrogenated in 90% acetic acid over rhodium on carbon to yield the decahydroisoquinoline (III) as a mixture of diastereoisomers. Treatment of (III) with benzyl chloroformate in aqueous sodium hydroxide solution gave a mixture of N-protected amino acids which was separated by fractional crystallization of the cyclohexylamine salts to give the (S,S,S)-isomer. Reaction with dicyclohexylcarbodiimide and N-hydroxysuccinimide in dimethoxyethane, followed by treatment of the activated ester with tert-butylamine in dichloromethane and subsequent hydrogenolysis of the benzyloxycarbonyl protecting group gave the decahydroisoquinoline (IV). In the other branch of the synthesis L-phenylalanine was treated with benzyl chloroformate in aqueous sodium hydroxide solution to give the N-protected amino acid. This was converted to the corresponding mixed anhydride with isobutyl chloroformate and N-ethylmorpholine in tetrahydrofuran and immediately reacted with diazomethane in diethyl ether to give the diazomethyl ketone (V). Treatment of (V) with ethereal hydrogen chloride gave the chloromethyl ketone (VI), which on reduction with sodium borohydride in aqueous tetrahydrofuran gave a mixture of diastereoisomeric chlorohydrins. Solvent extraction with boiling n-hexane followed by recrystallization of the less soluble isomer from isopropanol gave pure chlorohydrin (VII), which on treatment with ethanolic potassium hydroxide gave the epoxide (VIII). Condensation of (VIII) with (IV) in ethanol gave the hydroxyethylamine (IX). Hydrogenolysis of (IX) was followed by condensation with N-benzyloxycarbonyl-L-asparagine in tetrahydrofuran in the presence of 1-hydroxybenzotriazole and dicyclohexylcarbodiimide. Hydrogenolysis in ethanol over palladium on charcoal, followed by condensation with quinoline-2-carboxylic acid in tetrahydrofuran in the presence of dicyclohexylcarbodiimide and 1-hydroxybenzotriazole, gave the free base, Ro-31-8959/000. Treatment with methanesulfonic acid in aqueous ethanol then afforded the mesylate salt (X), Ro-31-8959/003.
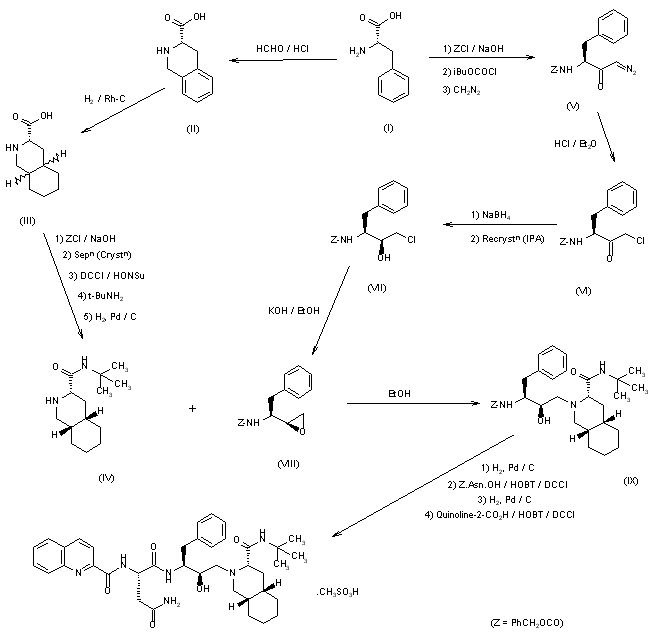
SYN
J Org Chem 1994,59(13),3656
Various new routes for the large-scale synthesis of Ro-31-8959 have been described: 1) The condensation of N-protected-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) gives the keto ester (III), which is enantioselectively reduced with NaBH4 to yield the hydroxy ester (IV). The reaction of (IV) with 2,2-dimethoxypropane (V) by means of p-toluenesulfonic acid affords the oxazolidine (VI), which is hydrolyzed with NaOH in ethanol/water to the corresponding acid (VII). The treatment of (VII) with oxalyl chloride, mercaptopyridine-N-oxide (MPO) and bromotrichloromethane affords the bromomethyloxazolidine (VIII), which, without isolation, is treated with acetic acid to give the N-protected 3(S)-amino-2-bromo-4-phenyl-2(S)-butanol (IX). The reaction of (IX) with KOH in methanol yields the epoxide (X), which is condensed with (3S,4aS,8aS)-N-tert-butyldecahydroisoquinoline-3-carboxamide (XI), yielding the protected condensation product (XII). The deprotection of the amino group of (XII) by hydrogenation with H2 over Pd/C affords the amino derivative (XIII), which is condensed with N-benzyloxycarbonyl-asparagine (XIV) in the usual way, giving the protected peptide (XV). The deprotection of (XV) as before yields compound (XVI), with a free amino group that is finally condensed with quinoline-2-carboxylic acid (XVII) by means of dicyclohexylcarbodiimide and hydroxybenzotriazole.
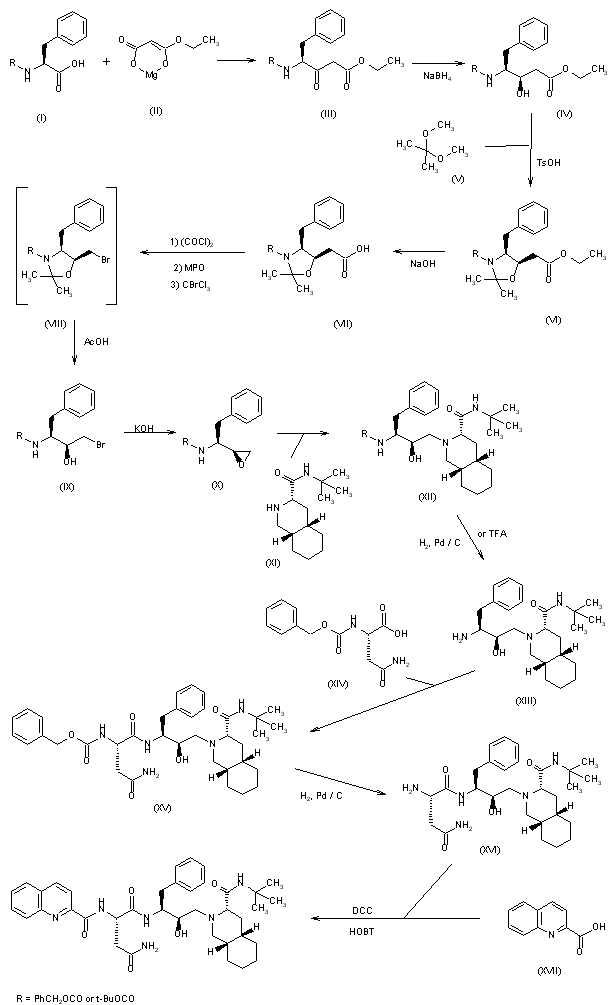
SYN
2) The condensation of N-phthaloyl-L-phenylalaninyl chloride (XVIII) with 1,1,2-tris(trimethylsilyloxy)ethylene (TMS) (XIX) at 90-100 C followed by acidic hydrolysis with HCl gives the acid (XX), which, without isolation, is decarboxylated, yielding 1-hydroxy-3(S)-phthalimido-4-phenyl-2-butanone (XXI). Sequential protection of the OH- group with dihydropyran, reduction of the CO group with NaBH4, mesylation of the resulting OH group with methanesulfonyl chloride and deprotection of the primary OH group gives 2(R)-(methanesulfonyloxy)-4-phenyl-3(S)-phthalimido-1-butanol (XXII). The epoxidation of (XXII) with potassium tert-butoxide yields the epoxide (XXIII), which is condensed with the decahydroisoquinoline (XI) as before, affording the protected condensation product (XXIV). The elimination of the phthalimido group of (XXIV) with methylamine and HCl gives the amino derivative (XIII), already obtained in scheme 16810301a.
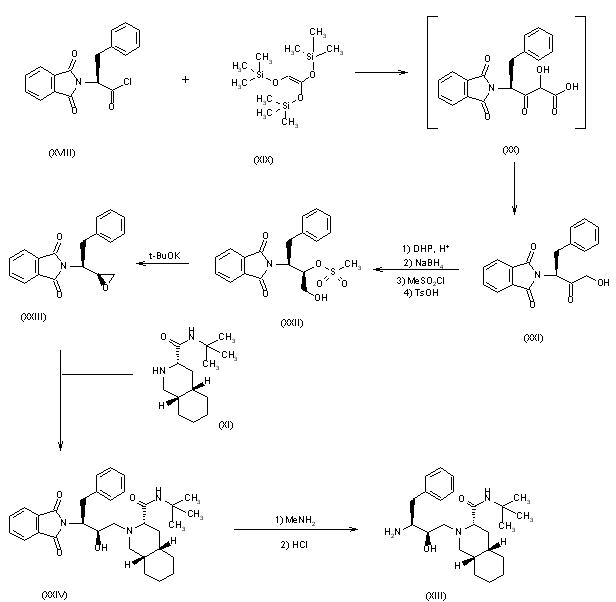
SYN
3) The condensation of N-(tert-butoxycarbonyl)-L-phenylalaninal (XXV) with 2-(trimethylsilyl)thiazole (XXVI) by means of tetrabutylammonium fluoride gives the thiazole derivative (XXVII), which is cleaved by reaction with methyl iodide (formation of the thiazolium derivative) and treated with NaBH4 and HgCl2 to afford the protected 3(S)-amino-2(S)-hydroxy-4-phenylbutanal (XXVIII). Finally, this compound is reductocondensed with isoquinoline (XI) by means of sodium cyanoborohydride to yield the protected condensation product (XII), already obtained in scheme 16810301a.
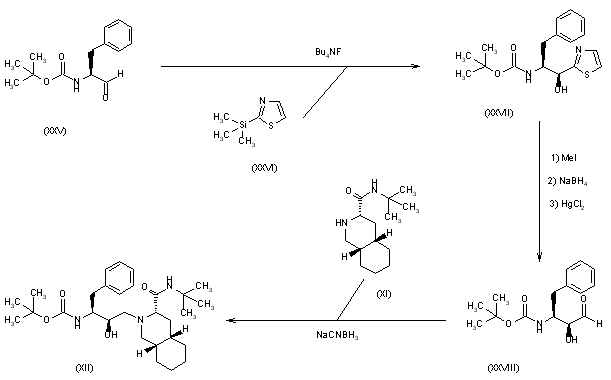
SYN
4) The selective esterification of 3(S)-azido-4-phenylbutane-1,2(S)-diol (XXIX) with 2,4,6-triiosopropylbenzenesulfonyl chloride (XXX) gives the sulfonate ester (XXXI), which by treatment with KOH is converted to the azido epoxide (XXXII). The condensation of (XXXII) with decahydroisoquinoline (XI) affords the azido condensation product (XXXIII), which is finally hydrogenated with H2 over Pd/C to the amino condensation product (XIII), already obtained in scheme 16810301a. 5) The reaction of (XXIX) with SOCl2 and RuCl3 gives the dioxathiole dioxide (XXXIV), which is condensed with decahydroisoquinoline (XI) to afford the azido condensation product (XXXIII), already obtained.
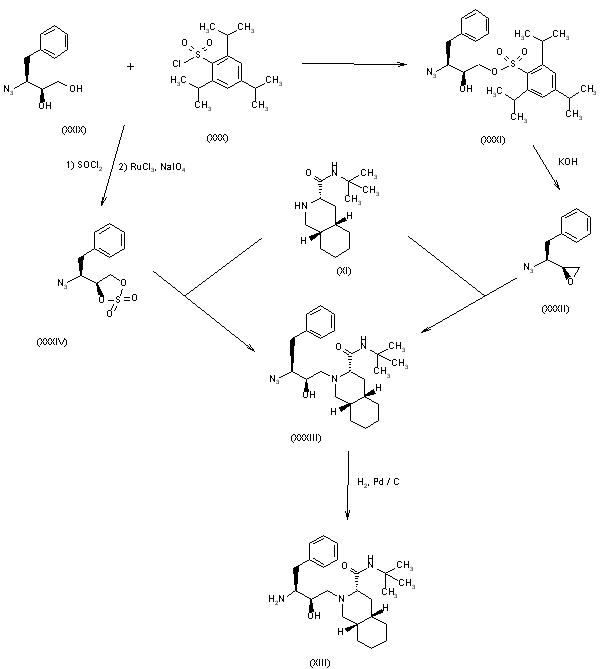
SYN
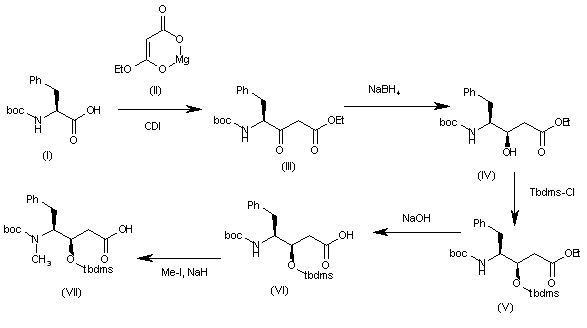
The intermediate (3R,4S)-4-[N-(tert-butoxycarbonyl)-N-methylamino]-5-phenyl-3-(tert-butyldimethylsilyloxy)pentanoic acid (VII) has been obtained as follows: The condensation of N-(tert-butoxycarbonyl)-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) by means of CDI gives the beta-ketoester (III), which is reduced with NaBH4 to yield (3R,4S)-4-(tert-butoxycarbonylamino)-3-hydroxy-5-phenylpentanoic acid ethyl ester (IV). The protection of the OH group of (IV) with Tbdms-Cl and imidazole in DMF affords the silylated ester (V), which is hydrolyzed with NaOH to provide the corresponding carboxylic acid (VI). Finally, this compound is N-methylated by means of Me-I and NaH in THF to obtain the target intermediate (VII).
SYN
J Label Compd Radiopharm 1998,41(12),1103
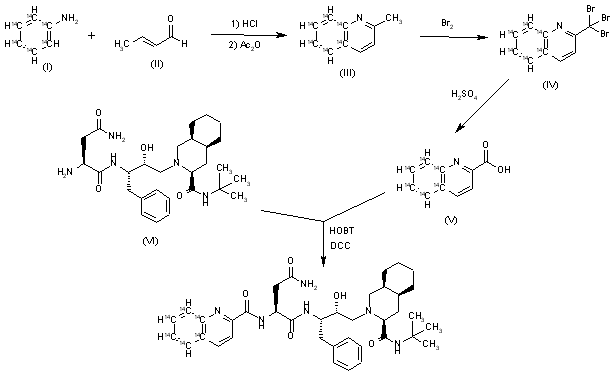
[14C]-Saquinavir: The cyclization of [ring-14C]-aniline (I) with crotonic aldehyde (II) by means of HCl and acetic anhydride gives labeled 2-methylquinoline (III), which is brominated with Br2 in acetic acid yielding the tribromo derivative (IV). The hydrolysis of (IV) with hot sulfuric acid afforded labeled quinoline-2-carboxylic acid (V), which is finally condensed with Ro-32-0445 (VI) by means of hydroxybenzotriazole (HOBT) and dicyclohexylcarbodiimide (DCC) in THF.
SYN
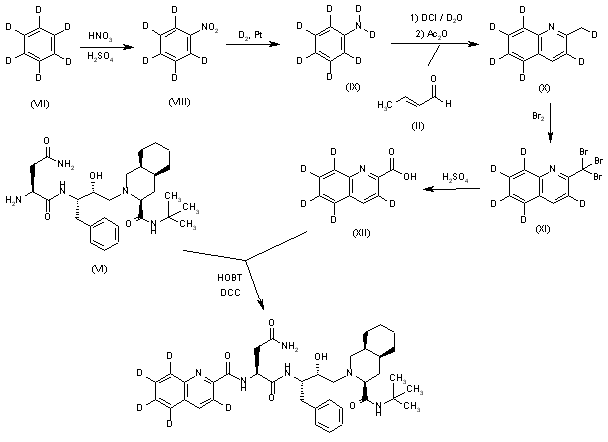
Pentadeuterated saquinavir: The nitration of hexadeuterobenzene (VII) with HNO3/H2SO4 gives pentadeuteronitrobenzene (VIII), which is hydrogenated with deuterium/Pt in D1-methanol yielding heptadeuteroaniline (IX). The cyclization of (IX) with crotonic aldehyde (II) by means of DCI/D2O and acetic anhydride as before affords hexadeuterated quinoline (X), which is brominated with Br2 as before giving the tribromo derivative (XI). The hydrolysis of (XI) with sulfuric acid as before yields the acid (XII), which is finally condensed with Ro-32-0445 (VI) as before.
SYN
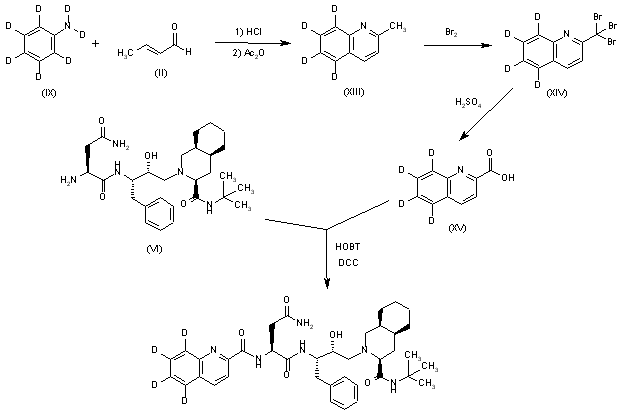
Tetradeuterated saquinavir: The cyclization of heptadeuteroaniline (IX) with crotonic aldehyde (II) by means of HCl and acetic anhydride as before gives the tetradeuteroquinoline (XIII), which is brominated as described yielding the tribromo derivative (XIV). The hydrolysis of (XIV) with sulfuric acid affords tetradeuterated acid (XV), which is finally condensed with Ro-32-0445 (VI) as indicated.
SYN
Tritiated saquinavir: The cyclization of 4-bromoaniline (XVI) with crotonic aldehyde (II) by means of ZnCl2/HCl gives 6-bromo-4-methylquinoline (XVII), which is brominated as before giving tetrabromo derivative (XVIII). The hydrolysis of (XVIII) with sulfuric cid affords 6-bromoquinoline-2-carboxylic acid (XIX), which is condensed with Ro-32-0445 (VI) by means of HOBT and DCC as indicated giving the bromo derivative of saquinavir (XX). Finally, this compound is tritiated with T2 over Pd/C in ethanol.
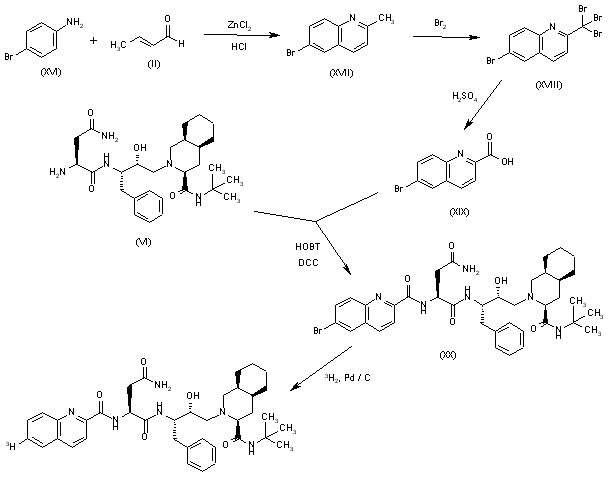
SYN
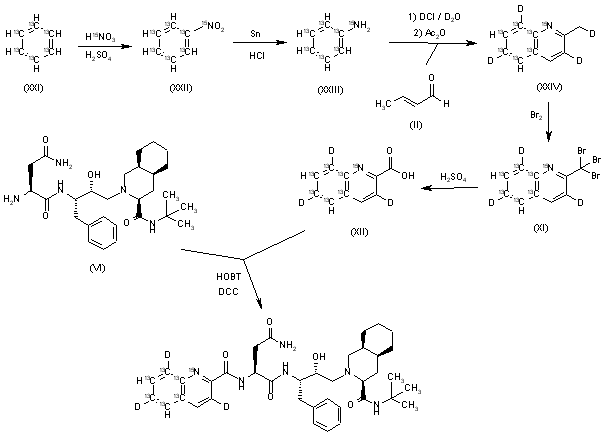
5)[15N,13C,2H]-Saquinavir: The nitration of [13C6]-benzene (XXI) with [15N]-nitric acid gives the corresponding nitrobenzene (XXII), which is reduced with Sn/HCl to the aniline (XXIII). The cyclization of (XXIII) with crotonic aldehyde (II) by means of ClD/D2O and acetic ahydride yields the tetradeuterated quinoline (XXIV), which is brominated as before givig the tribromo derivative (XXV). The hydrolysis of (XXV) with sulfuric acid as usual affords the [15N,13C6,2H3]-labeled quinoline-2-carboxylic acid (XXVI), which is finally condensed with Ro-32-0445 (VI) by means of HOBT and CDI as indicated.
Saquinavir (SQV), sold under the brand names Invirase and Fortovase, is an antiretroviral drug used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3] It is taken by mouth.[3]
Common side effects include nausea, vomiting, diarrhea, and feeling tired.[3] More serious side effects include problems with QT prolongation, heart block, high blood lipids, and liver problems.[3] It appears to be safe in pregnancy.[3] It is in the protease inhibitor class and works by blocking the HIV protease.[3]
Saquinavir was patented in 1988 and first sold in 1995.[4][5]
Medical uses[edit]
Saquinavir is used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3]
Side effects[edit]
The most frequent adverse events with saquinavir in either formulation are mild gastrointestinal symptoms, including diarrhoea, nausea, loose stools and abdominal discomfort. Invirase is better tolerated than Fortovase.[medical citation needed]
Bioavailability and drug interactions[edit]
Saquinavir, in the Invirase formulation, has a low and variable oral bioavailability, when given alone. The Fortovase formulation at the standard dosage delivers approximately eightfold more active drug than Invirase, also at the standard dosage.[6]
In the clinic, it was found that the oral bioavailability of saquinavir in both formulations significantly increases when patients also receive the PI ritonavir. For patients, this has the major benefit that they can take less saquinavir, while maintaining sufficient saquinavir blood plasma levels to efficiently suppress the replication of HIV.[medical citation needed]
The mechanism behind this welcome observation was not directly known, but later it was determined that ritonavir inhibits the cytochrome P450 3A4 isozyme. Normally, this enzyme metabolizes saquinavir to an inactive form, but with the ritonavir inhibiting this enzyme, the saquinavir blood plasma levels increased considerably. Additionally, ritonavir also inhibits multidrug transporters, although to a much lower extent.[medical citation needed]
Unlike other protease inhibitors, the absorption of saquinavir seems to be improved by omeprazole.[7]
Mechanism of action[edit]
Saquinavir is a protease inhibitor. Proteases are enzymes that cleave protein molecules into smaller fragments. HIV protease is vital for both viral replication within the cell and release of mature viral particles from an infected cell. Saquinavir binds to the active site of the viral protease and prevents cleavage of viral polyproteins, preventing maturation of the virus. Saquinavir inhibits both HIV-1 and HIV-2 proteases.[8]
History[edit]

New HIV infections and deaths, before and after the FDA approval of “highly active antiretroviral therapy”,[9] of which saquinavir, ritonavir and indinavir were key as the first three protease inhibitors.Cully, Megan (28 November 2018). “Protease inhibitors give wings to combination therapy”. nature. Open Publishing. Retrieved 28 October 2020. As a result of the new therapies, HIV deaths in the United States fell dramatically within two years.}}[9]
Saquinavir was developed by the pharmaceutical company Roche.[10] Saquinavir was the sixth antiretroviral and the first protease inhibitor approved by the US Food and Drug Administration (FDA), leading ritonavir and indinavir by a few months.[11] This new class of antiretrovirals played a critical role in the development of highly active antiretroviral therapy (HAART), which helped significantly lower the risk of death from AIDS-related causes, as seen by a reduction of the annual U.S. HIV-associated death rate, from over 50,000 to about 18,000 over a period of two years.[9][12]
Roche requested and received approval of Invirase via the FDA’s “Accelerated Approval” program—a process designed to speed drugs to market for the treatment of serious diseases—a decision that was controversial, as AIDS activists disagreed over the benefits of thorough testing versus early access to new drugs.[13][better source needed] It was approved again on November 7, 1997, as Fortovase,[14] a soft gel capsule reformulated for improved bioavailability. Roche announced in May 2005 that, given reduced demand, Fortovase would cease being marketed early in 2006, in favor of Invirase boosted with ritonavir,[15] owing to the ability of the latter co-formulated drug to inhibit the enzyme that metabolizes the AIDS drugs.[citation needed]
Society and culture[edit]
Economics[edit]
As of 2015, it is not available as a generic medication.[16]
Formulations[edit]
Two formulations have been marketed:
- a hard-gel capsule formulation of the mesylate, with trade name Invirase, which requires combination with ritonavir to increase the saquinavir bioavailability;
- a soft-gel capsule formulation of saquinavir (microemulsion,[17] orally-administered formulation), with trade name Fortovase, which was discontinued worldwide in 2006.[18]
References[edit]
- ^ “Saquinavir Use During Pregnancy”. Drugs.com. 20 March 2018. Retrieved 28 January 2020.
- ^ “Invirase- saquinavir mesylate capsule INVIRASE- saquinavir mesylate tablet, film coated”. DailyMed. 26 December 2019. Retrieved 28 January 2020.
- ^ Jump up to:a b c d e f g h i “Saquinavir”. The American Society of Health-System Pharmacists. Archived from the original on 8 September 2015. Retrieved 5 September 2015.
- ^ Minor, Lisa K. (2006). Handbook of Assay Development in Drug Discovery. Hoboken: CRC Press. p. 117. ISBN 9781420015706. Archived from the original on 31 March 2016.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 509. ISBN 9783527607495.
- ^ “Fortovase”. Drugs.com. 22 March 2019. Retrieved 28 January2020.
- ^ Winston A, Back D, Fletcher C, et al. (2006). “Effect of omeprazole on the pharmacokinetics of saquinavir-500 mg formulation with ritonavir in healthy male and female volunteers”. AIDS. 20 (10): 1401–6. doi:10.1097/01.aids.0000233573.41597.8a. PMID 16791014. S2CID 44506039.
- ^ Raphael Dolin, Henry Masur, Michael S. Saag. “AIDS Therapy“, Churchill Livingstone, (1999), p. 129.
- ^ Jump up to:a b c “HIV Surveillance—United States, 1981-2008”. Archivedfrom the original on 9 November 2013. Retrieved 8 November 2013.
- ^ J. Hilts, Philip (8 December 1995). “MF.D.A. Backs A New Drug To Fight AIDS”. New York Times. Retrieved 28 October 2020.
- ^ “Antiretroviral Drug Discovery and Development”. NIH. 26 November 2018. Retrieved 29 October 2020.
- ^ The CDC, in its Morbidity and Mortality Weekly Report, ascribes this to “highly active antiretroviral therapy”, without mention of either of these drugs, see the preceding citation. A further citation is needed to make this accurate connection between this drop and the introduction of the protease inhibitors.
- ^ “Drugs! Drugs! Drugs! An Overview of the Approved Anti-HIV Medications”. The Body. Archived from the original on 9 November 2013. Retrieved 20 February 2013.
- ^ “Drug Approval Package: Fortovase/Saquinavir NDA 20828”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 28 January 2020.
- ^ Withdrawal of Fortovase (PDF) Archived 2006-05-14 at the Wayback Machine
- ^ “Generic Invirase Availability”. Drugs.com. Retrieved 9 July2020.
- ^ Gibaud S, Attivi D (August 2012). “Microemulsions for oral administration and their therapeutic applications” (PDF). Expert Opinion on Drug Delivery. 9 (8): 937–51. doi:10.1517/17425247.2012.694865. PMID 22663249. S2CID 28468973.
- ^ News-Medical.Net. May 18, 2005 Roche to discontinue the sale and distribution of Fortovase (saquinavir) Archived 2015-02-22 at the Wayback Machine
External links[edit]
- “Saquinavir”. Drug Information Portal. U.S. National Library of Medicine.
links
| Clinical data | |
|---|---|
| Trade names | Invirase, Fortovase |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696001 |
| License data | EU EMA: by INNUS DailyMed: Saquinavir |
| Pregnancy category | AU: B1[1] |
| ATC code | J05AE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | ~4% (without ritonavir boosting)[2] |
| Protein binding | 98% |
| Metabolism | Liver, mainly by CYP3A4 |
| Elimination half-life | 9–15 hours |
| Excretion | feces (81%) and urine (3%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 127779-20-8 |
| PubChem CID | 441243 |
| IUPHAR/BPS | 4813 |
| DrugBank | DB01232 |
| ChemSpider | 390016 |
| UNII | L3JE09KZ2F |
| KEGG | D00429 |
| ChEMBL | ChEMBL114 |
| NIAID ChemDB | 000640 |
| PDB ligand | ROC (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6044012 |
| Chemical and physical data | |
| Formula | C38H50N6O5 |
| Molar mass | 670.855 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
///////////////saquinavir, Antiviral, Peptidomimetics, HIV Protease Inhibitor, Ro-31-8959, EU 2021, APPROVALS 2021, Invirase, Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
[H][C@@]12CCCC[C@]1([H])CN(C[C@@H](O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CC(N)=O)NC(=O)C1=NC3=C(C=CC=C3)C=C1)[C@@H](C2)C(=O)NC(C)(C)C
