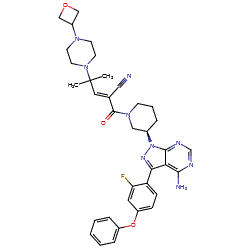
![(R)-2-(3-(4-Amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]-pyrimidin-1-yl)piperidine-1-carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin-1-yl)pent-2-enenitrile.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118325989&t=l)

PRN 1008, Rilzabrutinib
CAS 1575591-66-0
| リルザブルチニブ; |
C36H40FN9O3,
| MW 665.7597 |
2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-1-yl]pent-2-enenitrile
Anti-inflammatory disease, Autoimmune disease treatment
- OriginatorPrincipia Biopharma
- Class2 ring heterocyclic compounds; Amines; Anti-inflammatories; Fluorobenzenes; Nitriles; Phenyl ethers; Piperazines; Piperidines; Pyrazoles; Pyrimidines; Skin disorder therapies; Small molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase inhibitors
- Orphan Drug StatusYes – Idiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIIIdiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIAutoimmune disorders
- 02 Jun 2021Efficacy data from a phase IIa trial in Ankylosing spondylitis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
- 07 Apr 2021Sanofi initiates enrollment in a phase I pharmacokinetics trial in healthy volunteers in Australia (PO, Tablet, Capsule) (NCT04748926)
- 31 Mar 2021Sanofi announces intention to seek regulatory approval for Idiopathic thrombocytopenic purpura in 2023 (Sanofi pipeline, May 2021)
CLIP
Sanofi to acquire BTK inhibitor firm Principia for $3.7 billion
Principia is testing its small-molecule compounds in multiple sclerosis and immune system diseases
Sanofi will pay $3.7 billion to acquire Principia Biopharma, a San Francisco-based biotech firm developing small molecules that inhibit Bruton tyrosine kinase (BTK). The price represents about a 75% premium over Principia’s stock market value in early July, before reports surfaced that Sanofi was interested in buying the firm.
BTK is a protein important for both normal B cell development and the proliferation of lymphomas, which are B cell cancers. AbbVie, AstraZeneca, and BeiGene all market BTK inhibitors for treating specific kinds of lymphomas. Sales of AbbVie’s inhibitor, Imbruvica, approached $4.7 billion in 2019.
Other drug firms have been eager to get in on the action as well. In January, Merck & Co. spent $2.7 billion to acquire ArQule, whose experimental noncovalent BTK inhibitor is designed to overcome resistance that some cancers develop after treatment with current covalent BTK inhibitors. Eli Lilly and Company’s $8 billion acquisition of Loxo Oncology in 2019 also included a noncovalent BTK inhibitor.
BTK is also linked to inflammation, and Principia focuses on developing BTK inhibitors for immune system diseases and multiple sclerosis. Its compound rilzabrutinib is currently in clinical trials for pemphigus and immune thrombocytopenia. In 2017, Sanofi struck a deal to develop Principia’s brain-penetrant BTK inhibitor, SAR442168, for multiple sclerosis.
Sanofi announced in April of this year that the inhibitor reduced formation of new lesions—the scarred nervous tissue that gives multiple sclerosis its name—by 85% in a Phase II clinical trial. A Phase III trial of the compound began in June.
Upon announcing its deal to acquire Principia, Sanofi said that both rilzabrutinib and SAR442168 have the potential to become a “pipeline in a product,” indicating they can be used for many immune-related and neurological diseases, respectively.
The anti-inflammatory effects of BTK inhibitors have raised interest in the drugs as treatments for people hospitalized with COVID-19. Notably, the US National Cancer Institute conducted a small study suggesting acalabrutinib may help reduce the respiratory distress and inflammation in people with COVID-19. Based on that preliminary study, AstraZeneca—which markets acalabrutinib as Calquence—is conducting a 60-person randomized trial of the drug for COVID-19.
Sanofi has not indicated interest in investigating Principia’s BTK inhibitors as COVID-19 treatments.Chemical & Engineering NewsISSN 0009-2347
PATENTWO 2021127231https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127231&tab=PCTDESCRIPTION&_cid=P20-KRA0I9-18818-1
SOLID FORMS OF 2-[3-[4-AMTNO-3-(2-FT,TTORO-4-PHENOXY- PHEN¥L)PYRAZOLO[3,4 D]PYRIMIDIN l~YL]PIPERIDINE~l~CARBON¥L] 4~
METHYL-4-[4-(OXETAN-3-YL)PIPERAZIN-l-YLjPENT-2-ENENITRILE
[11 This application claims the benefit of priority to U.S. Provisional Application
No 62/951,958, filed December 20, 2019, and U.S Provisional Application No. 63/122,309, filed December 7, 2020, the contents of each of which are incorporated by reference herein in their entirety.
[2] Disclosed herein are solid forms of 2-[3-[4~amino-3~(2~fluoro-4-phenoxy-plienyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l Carbonyl]~4-nietliyl-4~[4-(oxetaii~3-yl)piperazin-!~yi]pent-2~enenitriie (Compound (I)), methods of using the same, and processes for making Compound (I), including its solid forms. The solid forms of Compound (I) may be inhibitors of Bruton’s tyrosine kinase (BTK) comprising low residual solvent content.
[3| The enzyme BTK is a member of the Tec family non-receptor tyrosine kinases.
BTK is expressed in most hematopoietic cells, including B cells, mast cells, and macrophages BTK plays a role in the development and activation of B cells. BTK activity has been implicated in the pathogenesis of several disorders and conditions, such as B cell-related hematological cancers (e.g., non-Hodgkin lymphoma and B cell chronic lymphocytic leukemia) and autoimmune diseases (e.g., rheumatoid arthritis, Sjogren’s syndrome, pemphigus, IBD, lupus, and asthma).
[4] Compound (I), pharmaceutically acceptable salts thereof, and solid forms of any of the foregoing may inhibit BTK and be useful in the treatment of disorders and conditions mediated by BTK activity. Compound (I) is disclosed in Example 31 of WO 2014/039899 and has the following structure:
where *C is a stereochemical center. An alternative procedure for producing Compound (!) is described in Example 1 of WO 2015/127310.
[5] Compound (I) obtained by the procedures described in WO 2014/039899 and WO 2015/127310 comprises residual solvent levels well above the limits described in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines. In general, manufacturing processes producing residual solvent levels near or above the ICH limits are not desirable for preparing active pharmaceutical ingredients (APIs).
Example 1: Spray Drying Process A
[311] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was washed with pH 3 phosphate buffer to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The dichloromethane solution was then washed with pH 7 buffer and solvent exchanged into isopropyl acetate. The isopropyl acetate solution was then washed with pH 3 phosphate buffer, bringing Compound (I) into the aqueous layer and removing non-basic impurities. The pH of the aqueous layer was adjusted to pH 9 with 10% sodium hydroxide, and the aqueous layer was extracted with isopropyl acetate. Upon concentration under vacuum, Compound (I) was precipitated from heptane at 0 °C, filtered and dried to give a white amorphous solid as a mixture of the (E) and (Z) isomers, as wet Compound (I). Wet Compound (I) was dissolved in methanol and spray dried at dryer inlet temperature of 125 °C to 155 °C and dryer outlet temperature of 48 to 58 °C to obtain the stable amorphous Compound (I) free base with levels of isopropyl acetate and heptane below 0.5% and 0.05%, respectively.
Example 2: Spray Drying Process B
intermediate A
Compound (!)
[241] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with Intermediate A (20.2 kg) and Intermediate B (13.6 kg, 1.5 equiv). DCM (361.3 kg, 14.5 vol) was charged to the reactor. The mixture was agitated, and the batch cooled to 0 °C to 5 °C. The reactor was charged with pyrrolidine (18.3 kg, 6 equiv) and then charged with TMSC1 (18.6 kg, 4 eq). Stirring was continued at 0 °C to 5 °C for 0.5 to 1 hour
[242] At 0 °C to 5 °C, acetic acid (2.0 equiv) was charged to the reactor followed by water (5 equiv). Stirring was continued at 0 °C to 5 °C for 1 to 1.5 hours. Water (10 equiv) was charged to the reactor, and the solution was adjusted to 20 °C to 25 °C. The internal temperature was adjusted to 20 °C to 25 °C and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[243] Water (7 vol) was charged to the reactor. The pH was adjusted to 2.8-3.3 with a 10 wt. % solution of citric acid. Stirring was continued at 0 to 5 °C for 1 to 1.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[244] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with an approximately 9% solution of NaHCCri (1 vol) and the organic layer. The internal temperature was adjusted to 20 °C to 25 °C, and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed. The aqueous layer was measured to have a pH greater than 7.
[245] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with the organic layer. The organic phase ¾s distilled under vacuum at less than 25 °C to 4 total volumes. IP AC (15 vol) was charged to the reactor. The organic phase was distilled under vacuum at less than 25 °C to 10 total volumes. Water (15 vol) followed by pH 2.3 phosphate buffer were charged to the reactor at an internal temperature of 20 °C to 25 °C. The pH adjusted to 3 Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed.
[246] The following steps were repeated twice: IP AC (5 vol) was charged to the reactor containing the aqueous layer. Stirring was continued for 0.25 to 0.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed. [247] IP AC (15 vol) was charged to the reactor containing the aqueous layer. A pH 10 phosphate buffer was charged to the reactor and the pH adjusted to 10 with 14% NaOH solution. Stirring was continued for 1.5 to 2 hours. Stirring was stopped and phases allowed to separate for at. least 0.5 h. The aqueous layer was discarded. The organic layer was dried over brine.
[248] The organic solution was distilled under vacuum at less than 25 °C to 5 total volumes.
[249] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with n-heptane (20 vol). The internal temperature was adjusted to 0 to 5 °C, and the IP AC solution was added.
[250] The suspension was filtered. The filter cake was washed with n-heptane and the tray was dried at 35 °C. Compound (I) (24.6 kg) was isolated in 86% yield.
[251] Compound (1) was dissolved in methanol (6 kg) and spray dried to remove residual IP AC and n-heptane.
Example 3: Precipitation Process A
[252] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (1) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the dichloromethane solution, and the dichloromethane solution was concentrated by distillation under reduced pressure, followed by addition of 1% NaCi aqueous solution and isopropyl acetate before adjustment of pH to approximately 3 with potassium hydroxide. The isopropyl acetate layer was removed and discarded. The aqueous layer containing Compound (I) was washed with isopropyl acetate to remove hydrophobic impurities. Washing was repeated as needed to reduce related substance impurities. Residual isopropyl acetate was removed by distillation under reduced pressure. The aqueous solution containing Compound (I) was cooled to 0 to 5°C before adjusting the pH to approximately 9 with potassium hydroxide. The free base of Compound (I) was allowed to precipitate and maturate at 20 °C for 20 hours. The mixture temperature was then adjusted to 20 °C to 25 °C, and the hydrate impurity was verified to be less than 0.3% (< 0.3%). The cake of the free base of Compound (I) was filtered and washed as needed to reduce conductivity. The cake was then allowed to dry on the filter under vacuum and nitrogen swept to reduce water content by Karl-Fischer (KF < 50%) before transferring to the oven for drying. The wet cake of the free base of Compound (1) was dried under vacuum at 25 °C until water content by Karl -Fischer was less than 1.5% (KF < 1.5%), and then dehmiped by milling to yield a uniform white amorphous solid as a mixture of the (E) and (Z) isomers, with no detectible levels of isopropyl acetate or heptane.
Example 4: Precipitation Process 3B
[253] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The washing was repeated as needed to reduce residual solvents and impurities. The dichloromethane solution was then washed with saturated sodium bicarbonate (pH > 7). Dichloromethane was removed by distillation under reduced pressure, followed by addition of water and isopropyl acetate. The pH of the aqueous layer was adjusted to pH to 2.8 – 3.3 with 2 M aqueous sulfuric acid (H2SQ4) at 0 – 5 °C, and the mixture rvas stirred and settled. After phase separation removal of the organic layer, the aqueous layer was washed with isopropyl acetate three times and the residual isopropyl acetate in aqueous layer was distilled out under vacuum at a temperature below 25 °C and the solution was basitied with 5% aqueous KOFI to pH 9 – 10 to a slurry . The resulting suspension was stirred and warmed up to 20 °C to 25 °C and aged for 20 h. The product was filtered and washed with water and dried to give white solid in 86% yield.
Example 5: Precipitation Process C
[254] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the d chloromethane solution, and the dichloromethane solution was concentrated under reduced pressure to obtain a thin oil. The concentrated oil was cooled to approximately 5°C before washing with an aqueous solution of sodium chloride. The organic phase was discarded. Washing of the aqueous layer was repeated as needed with dichloromethane to remove low level impurities. The pH of the aqueous solution was adjusted to approximately 3 with an aqueous solution of potassium hydroxide. Residual dichloromethane was removed
under reduced pressure. The level of residual acetic acid was determined by, for example, titration. The aqueous solution containing Compound (I) was cooled to a temperature between 0°C and 5°C. Acetic acid was present at 0 wt % to 8 wt. %. Acetic acid level was 0 wt % if the aqueous acid solution was washed with aqueous sodium bicarbonate or another aqueous inorganic base. Optionally, additional acetic acid was added to achieve a 0 wt.% to 8 wt. % acetic acid level. An aqueous solution of potassium hydroxide was constantly charged to the aqueous solution to obtain a pH to approximately 9.5. The free base of Compound (I) was allowed to precipitate and maturate at approximately 20 °C for least 3 hours. The cake (wet solid) of the free base of Compound (I) was filtered and washed with water. The wet cake was then dried under reduced vacuum with slight heat. Alternatively, instead of washing the wet cake with water, the wet cake was reslurried with water at approximately 15 °C for at least 1 hour before filtering. The free base of Compound (I) in the fomi of a wet cake was dried under vacuum with slight heat at 25°C.
[255] FIGs. 12-15 are example SEM images showing the variable morphologies of particles of Compound (I) during the filtration step to isolate Compound (I) based on the amount acetic acid added during the initial step in the precipitation of Compound (Ϊ) (FIG. 12: at 0 wt. % acetic acid; FIG 13: at 3 wt. % acetic acid; FIG. 14: at 5 wt. % acetic acid; FIG 15: at 8 wt. % acetic acid). Filtration speed depended on the morphology and was the fastest for 0 wt. % acetic acid. At 1 wt. % acetic acid, the filtration speed diminished considerably, improving at 2 wt. % to 3 wt. % acetic acid. Morphologies with more open holes (such as, e.g., more porous particles) resulted in improved filtration speeds, whereas more compact particles resulted in decreased filtration speed.
Example 6: Conversion of a Crystalline Form of Compound (Ϊ) to an Amorphous Form
[256] 9.8 grams of a crystalline form of Compound (I) were dissolved in approximately 20 mL of dichloromethane and approximately 120 ml. of brine solution. Then, approximately 1 equivalent of methanesulfonic acid was added. The pH w¾s approximately 2. The layers were separated. The aqueous layer was concentrated at a temperature between 0°C and 5°C to remove residual dichloromethane before slowly adding aqueous KOI I solution (approximately 5%) to adjust the pH to a value between 9 and 10. During aqueous KOH addition, an amorphous form of Compound (I) precipitated out. The slurry was slowly warmed to room temperature and then was stirred for approximately 24 hours before filtering and rinsing the wet cake with water. The wet cake was dried under vacuum with slight heat at approximately 30°C to provide 7 grams of a white to an off-white solid (87% yield and 98 4% purity). XRPD showed that the product was an amorphous solid form of Compound (I).
Example 7: Micronization of Compound (I) Particles Obtained by Precipitation Processes
[257] A fluid jet mill equipment was used during lab scale jet milling trials. The fluid jet mill equipment includes a flat cylindrical chamber with 1.5” diameter, fitted with four symmetric jet nozzles winch are tangentially positioned in the inner wall. Prior to feeding material to the fluid jet mill in each trial, the material was sieved in a 355 iim screen to remove any agglomerates and avoid blocking of the nozzles during the feed of material to the micronization chamber. The material to be processed was drawn into the grinding chamber through a vacuum created by the venturi (P vent ~ 0 5 – 1 0 bar above P grind). The feed flow rate of solids (F_feed) was controlled by a manual valve and an infinite screw volumetric feeder. Compressed nitrogen was used to inject the feed material; compressed nitrogen was also used for the jet nozzles in the walls of the milling chamber. Compressed fluid issuing from the nozzles expands from P grind and imparts very’ high rotational speeds in the chamber. Accordingly, material is accelerated by rotating and expanding gases and subjected to centrifugal forces. Particles move outward and are impacted by high velocity jets, directing the particles radially inward at very high speeds. Rapidly moving particles impact the slower moving path of particles circulating near the periphery of the chamber. Attrition takes place due to the violent impacts of particles against each other. Particles with reduced size resulting from this sequence of impacts are entrained in the circulating stream of gas and swept against the action of centrifugal force toward the outlet at the center. Larger particles in the gas stream are subjected to a centrifugal force and returned to the grinding zone. Fine particles are carried by the exhaust gas to the outlet and pass from the grinding chamber into a collector.
[258] The feeder has continuous feed rate control; however, to more precisely control the feed rate, the full scale of feed rates was arbitrary divided in 10 positions. To calibrate F feed, the feeder was disconnected from milling chamber and 10 g of Compound (I) powder was fed through the feeder operating at various feed rate positions. The mass of powder flowing through the feeder over 6 minutes was marked. The resulting feed rate was directly proportional to feeder position. After processing each of the four trials, the jet mill was stopped, micronized product removed from the container, and the milling chamber checked for any powder accumulation.
Variables/Parameters
F_feed Feed flow rate of solids [kg/h]
P grind Grinding pressure inside the
drying chamber [bar]
P vent Feed pressure in the venturi [bar]
Example 8: Residual Solvent Levels
[251] Retention of process solvents (/.<?., res dual solvents) depends on van der Waal s’ forces that are unique to and an inherent property of each molecule. Additionally, solvent retention depends how the API solid is formed, isolated, washed, and dried (i.e., during the manufacturing process). Because residual solvents may pose safety risks, pharmaceutical processes should be designed to minimize residual solvent levels (e.g , to result in residual solvent levels below the limits established in the ICH guidelines).
[252] Residual solvent analysis was performed using gas chromatography-mass spectrometry. The residual solvent levels in solid forms of Compound (I) prepared by spray drying processes described herein and precipitation processes described herein are provided in Table 2. The residual solvent levels in crude Compound (I) listed in Table 2 are comparable to the residual solvent levels in crude Compound (I) prepared according to the procedures detailed in Example 31 of WO 2014/039899 and Example 1 of WO 2015/127310.
Table 2: Residual solvent levels in solid forms of Compound (I)
PATENTWO 2015127310https://patents.google.com/patent/WO2015127310A1/enExample 1Synthesis of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l- yl]-piperidine-l-carbonyl]-4-m iperazin-l-yl]pent-2-enenitrile

Step 1To a solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l -yl]-l-piperidyl]-3-oxo-propanenitrile (15 g, 3.12mmol), 2-methyl-2-[4- (oxetan-3-yl)piperazin-l-yl]propanal (794.25mg, 3.74mmol) in DCM (40mL), pyrrolidine (1.54mL,18.71mmol) at 0-5 °C was added, which is followed by TMS-Cl (1.58mL,12.47mmol). The reaction mixture was stirred at 0-5 °C for 3 h and was quenched with 1 M potassium phosphate buffer (pH 3). Layers were separated and the organic layer was washed once more with 1 M potassium phosphate buffer (pH 3). The organic layer was extracted withl M potassium Phosphate buffer at pH 1.5. Layers were separated. The aqueous phase contained the desired product while the impurities stayed in the organic phase. The aqueous phase was neutralized with 1 M potassium phosphate (pH 7) and was extracted with isopropylacetate (10 volumes). Upon concentration 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile was obtained as a foam having >99% HPLC purity. MS (pos. ion) m/z: 666 (M+l ).The foam containing high levels of residual solvent was dissolved in 2 M HC1 and the resulting solution was placed under vacuum to remove residual organic solvents. pH of the solution was then adjusted to ~ 7 and the resulting paste was filtered and dried in vacuum without heat. This resulted in isolation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3- yl)piperazin- l-yl]pent-2-enenitrile containing residual water up to 10%. Drying under vacuum without heat reduces the water level but lead to generation of impurities.Step 1AAlternatively, the isopropylacetate solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4- (oxetan-3-yl)piperazin-l -yl]pent-2-enenitrile can be concentrated to 4 vol and added to heptane (20 volume) at 0 °C. The resulting suspension was stirred at 0 °C overnight and the product was filtered, washed twice with heptane and dried at 45 °C for 2 days under vacuum to give 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l – yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile in 85 – 90 % yield as a free flowing solid. However, the solids obtained by this method contained high residual solvents (3.9 wt% isopropylacetate and 1.7 wt% heptane). In addition, the free base form was not very stable as degradation products were observed during the drying process at less than 45 °C.Salt formationExample 2Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4-d]pyrimidin- l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2-enenitrile hemisulfate and sulfate saltHemisulfate: To the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (4.2 g) in EtOAc (60 mL, 15 vol) was added sulfuric acid (0.31 g, 0.17 mL, 0.5 eq) in EtOAc (20 mL, 5 vol) at ambient temperature. The suspension was stirred at ambient temperature for ~ 2 hr and then 40 °C for 4 hr and then at ambient temperature for at least 1 hr. After filtration and drying at ambient temperature under vacuum, 1.5 g of white powder was obtained. Solubility of the hemi-sulfate at ambient temperature was > 100 mg/mL in water.Sulfate saltTo the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (810 mg) in EtOAc (8 mL, 10 vol) was added sulfuric acid (0.06 mL, 1.0 equiv.) in EtOAc (2.5 mL, 5 vol) at ambient temperature. The resulting suspension was stirred at 40 °C for 2 hr and then cooled to ambient temperature for at least 1 hr. After filtration, solids were dried by suction under Argon for 1 h to give a white powder (0.68 g) in 69% yield.
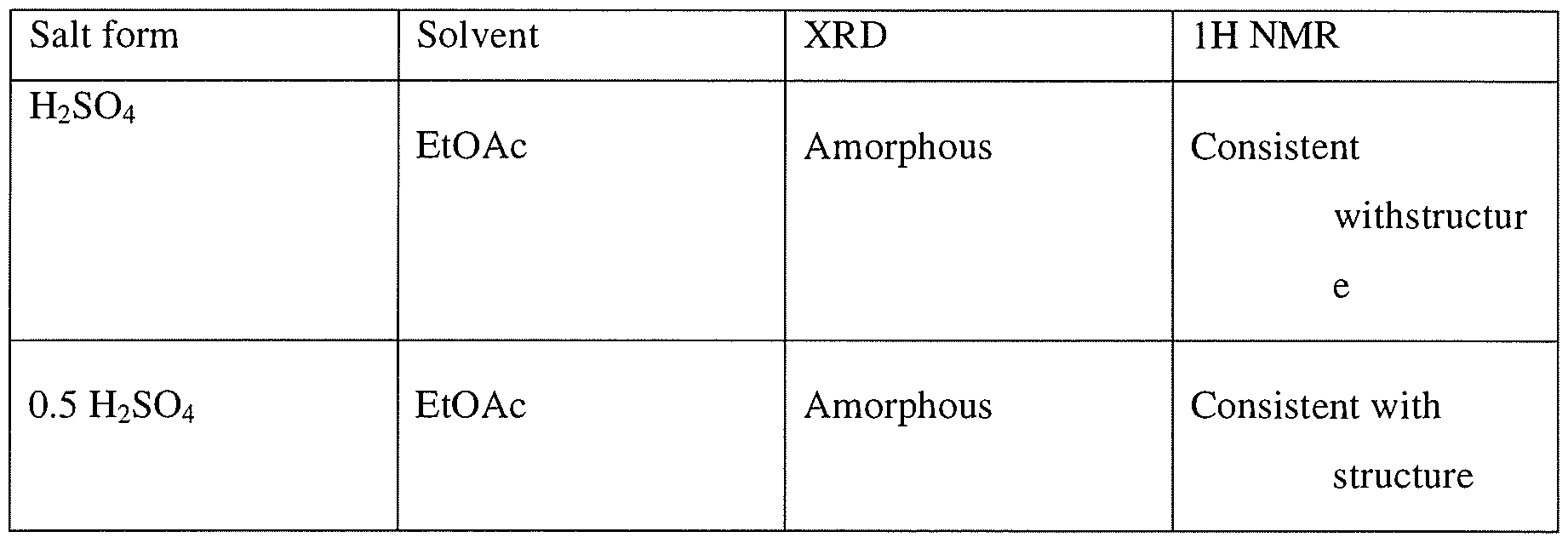
Example 3Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin- 1 -yl]-piperidine- 1 -carbonyl] -4-methyl-4-[4-(oxetan-3-yl)-piperazin- 1 -yl]pent-2- enenitrile hydrochlorideTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2CI2 (1ml) at ambient temperature was added 2 equivalent of HC1 (0.3 mmol, 0.15 ml of 2M HC1 in 1 : 1 dioaxane:CH2Cl2). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 15 volumes of ethylacetate (as compared to CH2C12) resulting in formation of a white solid. The mixtures was aged at ambient temperature for lh and placed at 2-8 C for 19 h. Upon filtration and washing of the filter cake with ethylacetate and drying a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt. IC indicated formation mono-HCl salt.

Example 4General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile mono- and di-mesylate saltsTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2C12 (1 ml) at ambient temperature was added either 1 equivalent of methanesulfonic acid (0.15 mmol, 0.2 ml of 74 mg/ml solution in CH2C12) or 2 equivalent of methanesulfonic acid (0.3 mmol, 0.4 ml of 74 mg/ml solution in CH2C12). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 10 volumes of antisolvents (ethylacetate, methyl tert-butylether (MTBE), or cyclohexane) (10 ml as compared to CH2C12) resulting in formation of a white solid. The mixture was aged at ambient temperature for lh and placed at 2-8 °C for 19 h. Upon filtration and washing of the filter cake with the antisolvent and drying, a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt as well as counterion ratio.Alternatively 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile can be dissolved in 4 volumes of isopropylacetate and added to 2 equivalent of methanesulfonic acid in 6 volumes of isopropylacetate at 0 °C to generate the dimesylate salt.

1. Theoretical mesylate content, monomesylate=12.6% and dimesylate=22.4%, NO- not determinedExample 5 General procedure for the preparation of carboxylate salt Approximately 20 mg of the compound (I) was dissolved in minimum amount of the allocated solvent system. These were then mixed with the appropriate number of equivalents of counterion dissolved or slurried in the allocated solvent.If compound (I) was insoluble in the selected solvent, slurry of the sample was used after adding 300 μί.If the acid was insoluble in the selected solvent, slurry of the acid was used after adding 300 xL.If the acid was a liquid, the acid was added to the dissolved/slurried compound (I) from a stock solution in the allocated solvent.The suspensions/ precipitates resulting from the mixtures of compound (I) were temperature cycled between ambient (ca. 22°C) and 40°C in 4 hour cycles for ca. 48 hrs (the cooling/heating rate after each 4 hour period was ca. 1 °C/min). The mixtures were visually checked and any solids present were isolated and allowed to dry at ambient conditions prior to analysis. Where no solid was present, samples were allowed to evaporate at ambient. Samples which produced amorphous material, after the treatment outlined above, were re- dissolved and precipitated using anti-solvent (ter/-butylmethylether) addition methods at ambient conditions (ca. 22°C). i.e. the selected anti-solvent was added to each solution, until no further precipitation could be observed visually or until no more anti-solvent could be added. The solvents used in this preparation were acetonitrile, acetone, isopropyl acetate, THF and MTBE. The acid used were oxalic acid, L-aspartic acid, maleic acid, malonic acid, L-tartaric acid, and fumaric acid.Example 6General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile hemicitrate saltTo a solution 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (5 g, 7.5 mmol) in ethanol (50 ml) was added citric acid (720.5 mg, 3.76 mmol) dissolved in 2 ml of water. Mixture was stirred at ambient temperature for 15 min, additional 0.5 ml of water was added and the mixture was stirred for 1 h, concentrated in vacuo to a gum. Ethanol was added and the mixture was concentrated. This process was repeated twice more and then CH2CI2 was added to the mixture. Upon concentration a white solid was obtained which was tumble dried under reduced pressure at 40 C for 4 h, then in a vacuum oven for 19h to give 5.4 g of a solid. Analysis by XRD indicated formation of an amorphous solid
PATENT
WO2014039899, Example 31
Rilzabrutinib (PRN1008) is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase (BTK) [1].
https://patents.google.com/patent/WO2014039899A1/enExample 31Synthesis of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)- 1 H-pyrazolo[3,4-d]pyrimidin- 1 -yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile

Step 1A solution of 2-bromo-2-methyl-propanal (696.6 mg, 4.61 mmol) in DCM (10 mL) was cooled with an ice bath and l -(oxetan-3-yl)piperazine (328 mg, 2.31 mmol), diluted with 5-10 mL of DCM, was slowly added via addition funnel over a 15 min period. Next, Hunig’s base (0.4 mL, 2.31 mmol) was added and then the cooling bath was removed. The reaction mixture was stirred at room temperature overnight and the DCM layer was washed three times with 0.5N HC1. The combined aqueous layer was neutralized with NaOH to pH 10-11 and extracted with DCM. The combined organic layer was washed with brine and dried over Na?S04. Filtration and removal of solvent afforded 2-methyl-2-[4-(oxetan-3-yl)piperazin-l- yl]propanal as a light yellow liquid, which was used directly in the next step without further purification.Step 2To a cooled (0 °C) solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)- pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidyl]-3-oxo-propanenitrile (80 mg, 0.17 mmol), was added 2-methyl-2-[4-(oxetan-3-yl)piperazin-l-yl]propanal (-108 mg, 0.51 mmol) in DCM (10 mL) followed by pyrrolidine (0.08 mL, 1.02 mmol) and TMS-C1 (0.09 raL, 0.68 mmol.) The ice bath was removed, and the reaction stirred 1 hour. Most of the solvent was removed and the residues were purified by chromatography, using 95:5 CH2Cl2:MeOH to obtain 79 mg of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)-lH-pyrazolo[3,4-d]-pyrimidin-l- yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile as a white solid. MS (pos. ion) m/z: 666 (M+l).
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0223523421001781?dgcid=rss_sd_all
Therapy based on Bruton’s tyrosine kinase (BTK) inhibitors one of the major treatment options currently recommended for lymphoma patients. The first generation of BTK inhibitor, Ibrutinib, achieved remarkable progress in the treatment of B-cell malignancies, but still has problems with drug-resistance or off-target induced serious side effects. Therefore, numerous new BTK inhibitors were developed to address this unmet medical need. In parallel, the effect of BTK inhibitors against immune-related diseases has been evaluated in clinical trials. This review summarizes recent progress in the research and development of BTK inhibitors, with a focus on structural characteristics and structure-activity relationships. The structure-refinement process of representative pharmacophores as well as their effects on binding affinity, biological activity and pharmacokinetics profiles were analyzed. The advantages and disadvantages of reversible/irreversible BTK inhibitors and their potential implications were discussed to provide a reference for the rational design and development of novel potent BTK inhibitors.

///////////////PRN-1008, PRN 1008, Rilzabrutinib, リルザブルチニブ,
N#CC(=CC(N(C1COC1)C)(C)C)C(=O)N1CCCC1Cn1nc(c2c1ncnc2N)c1ccc(cc1F)Oc1ccccc1