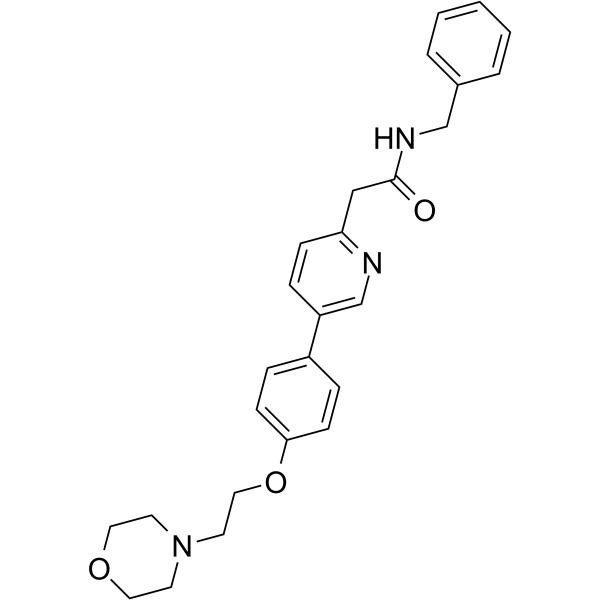
Tirbanibulin
CAS 897016-82-9, 1038395-65-1 DI HCL
1080645-95-9 MESYLATE
N-benzyl-2-[5-[4-(2-morpholin-4-ylethoxy)phenyl]pyridin-2-yl]acetamide
| Molecular Weight | 431.53 |
|---|---|
| Formula | C₂₆H₂₉N₃O₃ |
FDA APPROVED 12/14/2020, Klisyri
To treat actinic Keratosis of the face or scalp
Tirbanibulin (KX2-391) is an inhibitor of Src that targets the peptide substrate site of Src, with GI50 of 9-60 nM in cancer cell lines.
- Originator Kinex Pharmaceuticals
- Developer Almirall S.A.; Athenex; Hanmi Pharmaceutical; Kinex Pharmaceuticals; PharmaEssentia Corporation
- ClassAcetamides; Amides; Antineoplastics; Antipsoriatics; Morpholines; Phenyl ethers; Pyridines; Skin disorder therapies; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Src-Family kinase inhibitors; Tubulin polymerisation inhibitors
- PreregistrationActinic keratosis
- Phase IIPsoriasis
- Phase I/IISolid tumours
- Phase IPhotodamage
- PreclinicalSkin cancer
- 09 Mar 2020FDA assigns PDUFA action date of 30/12/2020 for tirbanibulin for Actinic keratosis
- 09 Mar 2020US FDA accepts NDA for tirbanibulin for Actinic keratosis for review
- 02 Mar 2020European Medicines Agency accepts Marketing Authorization Application for tirbanibulin for Actinic keratosis for review
KX-01 is a dual inhibitor of Src kinase and tubulin polymerization. KX01 promotes the induction of p53, G2/M arrest of proliferating cell populations and subsequent apoptosis via the stimulation of Caspase-3 and PARP cleavage. The drug was developed by Kinex Pharmaceuticals and reached phase II of clinical trials for the treatment of Castration-Resistant Prostate Cancer and Actinic Keratosis. KX-01 demonstrated good in vitro pofile against different cancer cell lines with IC50 in nanomolar range.
Tirbanibulin (Mesylate) (KX2-391 (Mesylate)) is an inhibitor of Src that targets the peptide substrate site of Src, with GI50 of 9-60 nM in cancer cell lines.
Tirbanibulin (KX2-391) is a Src inhibitor that is directed to the Src substrate pocket. Tirbanibulin (KX2-391) shows steep dose-response curves against Huh7 (GI50=9 nM), PLC/PRF/5 (GI50=13 nM), Hep3B (GI50=26 nM), and HepG2 (GI50=60 nM), four hepatic cell cancer (HCC) cell lines[1]. Tirbanibulin (KX2-391) is found to inhibit certain leukemia cells that are resistant to current commercially available drugs, such as those derived from chronic leukemia cells with the T3151 mutation. Tirbanibulin (KX2-391) is evaluated in engineered Src driven cell growth assays inNIH3T3/c-Src527F and SYF/c-Src527F cells and exhibits GI50 with 23 nM and 39 nM, respectively[2].
Orally administered Tirbanibulin (KX2-391) is shown to inhibit primary tumor growth and to suppress metastasis, in pre-clinical animal models of cancer[2].
[1]. Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci, 2009, 54(7), 1465-1474. [2]. Fallah-Tafti A, et al. Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities. Eur J Med Chem, 2011, 46(10), 4853-4858.

Approval allows Almirall to move forward with the topical ointment for individuals with AK on the face or scalp.

The US Food and Drug Administration (FDA) has approved tirbanibulin (Klisyri) as a topical treatment for actinic keratosis (AK).
The approval, awarded to Almirall, S.A., will allow the novel, topical first-in-class microtubule inhibitor for treatment of the disease on the face or scalp, representing a significant breakthrough in treatment of AK because of its short treatment protocol of once daily application for 5 days.
Actinic keratosis represents the second most common diagnosis in dermatology in the US, with a reported prevalence between 11-25%.
“Early diagnosis and treatment of actinic keratosis (AK) is critical, since those who already have an AK are likely to develop more actinic keratoses (plural) in the future,” said Deborah S. Sarnoff, MD, President of the Skin Cancer Foundation, said in a statement. “Patients with AK are at higher risk for skin cancer, since AKs can progress into squamous cell carcinoma (SCC), a common and sometimes invasive form of skin cancer.”
The approval is based on recent data from a large phase 3 clinical study, as well as 2 randomized, double-blind, vehicle-controlled phase 3 studies evaluating the efficacy and safety of tirbanibulin ointment 1% in adults with AK on the face or scalp.
“These studies enrolled a total of 702 patients across 62 sites in the United States, providing robust data,” Andrew Blauvelt, MD, MBA, President of Oregon Medical Research Center, and one of the lead investigators of the studies, said in a statement. “Tirbanibulin achieved a significantly higher number of patients with complete (100%) clearance of AK lesions in the treated area compared to vehicle (44% vs. 5% in study 1 and 54% vs. 13% in study 2), as well as reaching the secondary endpoint of partial (≥75%) clearance of lesions.”
PATENT
WO 2006071960
US 20070015752
US 20080287436
WO 2008082637
WO 2008002676
US 20090318450
https://patents.google.com/patent/US20090318450A1/en
- .
- [0374]A 1 L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165° C. oil bath temperature; internal temperature was 147° C.) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2×300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
- [0375]1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M+1].
- [0376]To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2×150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of ˜200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ˜1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ˜45° C., the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (˜45° C.) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (˜45° C.) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
- [0377]1H NMR (DMSO-d6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M+1 of free base].
- [0378]Elemental analysis (for C26H29N3O3.2 HCl.0.035 EtOH.0.29.EtOAc.0.03 heptane.0.8 H2O):
- [0379]Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91
- [0380]Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24
- [0381]Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
- [0382]The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
- [0383]Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
- [0384]A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
- [0385]The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (˜40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) of EtOAc].
PATENT
WO 2009051848
https://patents.google.com/patent/WO2009051848A1/en
].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I) free base).
[ 000242 ] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 0C oil bath temperature; internal temperature was 147 0C) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1:1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1:2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (>98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors.
[000243 ] 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M + I].

Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (Compound (I), diHCI salt).
[000244 ] To a stirred suspension of compound (I) (free base, 60.00 g) in absolute EtOH (600 mL) was added 170 mL of 2.5 M HCl (in ethanol), 25 mL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at -45 0C, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 0C) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (-45 0C) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
[000245] 1H NMR (DMSO-Cl6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M + 1 of free base].
[000246] Elemental analysis (for C26H29N3O3 • 2 HCl • 0.035 EtOH • 0.29 EtOAc • 0.03 heptane • 0.8 H2O): a. Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91 b. Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24
[ 000247] Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O).
[ 000248] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane.
[000249] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000250] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture:
[000251] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of compound (I) as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) ofEtOAc].
Example 3: Large Scale Synthesis of Compound (I) di-HCl
[000252 ] Reagents and solvents were used as received from commercial suppliers. Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra. Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2)
[000253 ] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 0C and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 °C. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 0C, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000254] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between —35 to -50 0C. While maintaining a temperature between -35 to —45 0C, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 0C. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 0C and 0 0C. The reaction mixture was stirred at 18 to 23 0C for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 * 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) [000255] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), 4- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 0C and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22°C. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 0C. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000256] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[ 000257] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 0C. While maintaining a temperature below -10 0C, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below —100C. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 0C. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (I x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 * 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 mL, 7 vol.) was added to the oil at 50 0C and stirred for 15 minutes. Heptane (850 mL, 5vol.) was added over ten minutes at 50 0C. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 0C to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000258] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 0C. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 0C. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 0C. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 0C. While maintaining a temperature below 20 0C, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[ 000259] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 0C and stirred for two days. The reaction mixture was cooled to 45-50 0C and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4: 1 Anisole/n-heptane (200 mL) and n-heptane (3 x 100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (Compound (I) 2HC1)
[000260 ] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 raL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to compound (I) (100 g) over 30 minutes while maintaining a temperature of 30 0C. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC).
Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
Crystallization Study of Compound (I) 2HCl
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (Compound (I))
[000268] To a 22-L reactor was charged compound 7 (650 g, 1.82 mol), anisole (3.25
L, 5 vol, anhydrous) and benzylamine (600 mL, 0.92 vol, 3 equiv). The batch (approximately 18 °C) was heated to 142 ± 5 °C over 1 hour 44 minutes, with dissolution occurring at 30 0C. The batch was maintained at 142 ± 5 0C for 69 hours 30 minutes at which point HPLC analysis indicated that compound 7 was 0.9% by conversion (specification <1.7% by conversion). The batch was cooled to 45-50 0C over 5 hours 12 minutes (to aid cooling the nitrogen flow was increased once the batch was approximately 72 0C). At that temperature range, the batch was poorly stirring and on mixing, the batch temperature increased to 52 0C. It was >50 °C for <15 minutes. The batch was aged for 2 hours 2 minutes once initially <50 0C, then n-heptane (9.75 L, 15 vol, 99%) was added to the batch over 1 hour 56 minutes, maintaining the batch temperature at 45-50 °C. The heating was then discontinued and the batch cooled to 25 0C over 10 hours 32 minutes and then to approximately 20 °C over 20 minutes. The total time the batch was maintained <25 0C was 4 hours 50 minutes (2 hours 47 minutes at approximately 20 0C). The batch was filtered under suction via a 24-inch polypropylene filter funnel (fitted with a PTFE cloth) and the reactor rinsed with anisole/n- heptane (1.3 L, 4: 1) and the rinse transferred to the cake. The cake was then washed successively with two portions of /i-heptane (1.3 L, 0.65 L). The total filtration time was 39 minutes. The batch (net wet weight 1004 g of KX2391) was transferred to three glass trays and placed into a vacuum oven set at 50 0C and dried to constant weight over 96 hours 26 minutes.
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (Compound (I)-MSA)
[000269] Compound (I) (520 g, 1.21 mol) was transferred to reactor 1 using acetone (41.6 vol, 80 vol, ACS) to facilitate the transfer. The batch was heated to 50 ± 5 0C over 33 minutes with dissolution occurring at 30 0C . The batch was clarified into a second reactor via a transfer pump fitted with an inline filter (Pall P/N 12077, 10 micron) and reheated from 46 0C to 50 ± 5 0C. Methanesulfonic acid (121.4 g, 1.05 equiv, 99% extra pure) was added to the pale yellow batch over 12 minutes and the heating then discontinued. After fourteen minutes, white solids were observed, which increased in number to give after 59 minutes a white suspension. The batch was in the range of 25 ± 5 0C after 7 hours 51 minutes and aged for a further 19 hours 21 minutes (10 hours 30 minutes at <27 0C). The batch was filtered under suction via a 24-inch polypropylene filter (PTFE cloth) and the reactor rinsed with acetone (2.0 L, clarified, ACS) and the rinse transferred to the cake. The cake was covered with a stainless steel cover and sucked dry under a flow of nitrogen. The total filtration time was 21 minutes. The batch (net wet weight 764 g) was transferred to three glass drying trays and dried in a vacuum oven to constant weight at 25 ± 5 °C over 21 hours 54 minutes (565 g, 89% of theory). A sample was removed for analysis and the batch maintained in vacuo at 25 ± 5 °C. The batch was then transferred to two 80-oz amber glass bottles (Teflon lined polypropylene closure), blanketed with argon and stored at -10 to -20 °C.
PATENT
WO 2010135429
https://patents.google.com/patent/WO2010135429A2/en
Preparation of KX2-391 and its salts
[00045] The synthesis of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine is shown in the scheme below:

[00046] 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was synthesized in 3 steps. Intermediate 2 was synthesized using an ether coupling reaction e.g., using Williamson ether synthesis. Ether formation between 4-(2-chloroethyl)morpholine (1) and A- bromophenol was carried out in the presence of potassium carbonate and DMF to afford 4-(2- (4-bromophenoxy)ethyl)morpholine (2). Rigorously dry conditions were not essential for this reaction and a basic wash with sodium hydroxide was used to remove any remaining A- bromophenol. In another aspect of the invention, intermediate 2 is synthesized using any ether formation reaction. Intermediate 2 is synthesized starting from compound 1 containing any leaving group. For example, the skilled chemist would start with compounds of the
general formula

wherein the leaving group “LG” includes but is not limited to halogen, tosylate, mesylate, trifluate, etc.
[00047] Compound 5 was formed using a Suzuki reaction. Formation of the aryl borate, 6-fluoropyridin-3-yl-3-boronic acid (4), was carried out by forming the aryl anion using n-BuLi followed by in situ quenching with triisopropylborate (Li, et ah, J. Org. Chem. 2002, 67, 5394-5397). The resulting 6-fluoropyridin-3-yl-3-boronic acid (4) was coupled to 4-(2-(4-bromophenoxy)ethyl)morpholine (2) in a solution of DME and aqueous sodium carbonate using tetrakis(triphenylphosphine)palladium to afford 4-(2-(4-(6-fluoropyridin-3- yl)phenoxy)ethyl)morpholine (5), which was purified using silica gel chromatography. The skilled chemist would know that other transition metal coupling reaction are used to prepare compound 5.
[00048] The synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide dihydro chloride is shown below:

[00049] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride (KX2-391 HCl) was synthesized in four linear steps. The fluoride of 4-(2-(4- (6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5) was displaced by the anion of acetonitrile formed using commercially available NaHMDS. Acetonitrile was added slowly to a cooled mixture of compound 5 and base to form 2-(5-(4-(2- morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6). In another aspect of the invention, intermediate 5 may have a leaving group other than fluorine. Thus, compounds of the general formula:

would be pursued where LG includes other leaving groups known to the skilled chemist.
[00050] Acid catalyzed methanolysis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-
2-yl)acetonitrile (6) was carried out using a mixture of concentrated sulfuric and fuming sulfuric acid. The use of fuming sulfuric acid removed residual water from the reaction mixture and reduced the amount of carboxylic acid by-product formed. The reaction mixture was quenched by adding the reaction mixture to a solution of saturated sodium bicarbonate and dichloromethane while maintaining the temperature below 20 ºC. Any carboxylic acid contaminant was readily removed with aqueous work-up. In another aspect of the invention, other acid catalyzed conditions are used by the skilled artisan for alcoho lysis of the nitrile of compound 6 to produce compound 7.
[00051] The resulting methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2- yl)acetate (7) and benzyl amine were coupled in anisole at high temperature to afford 2-(5-(4- (2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391). An HCl solution formed by adding acetyl chloride to absolute ethanol was added to KX2-391 to form the bis- HCl salt, 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride, (KX2-di-HCl).
[00052] The synthesis of the mesylate salt of KX2-391 (KX2-391 -MSA) is depicted in the scheme below:

[00053] 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide mesylate (KX2-391 MSA) was synthesized in four linear steps starting from compound 5.
The first 3 steps were carried out similar to the procedure discussed above for KX2-391 2HCl to afford methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (KX2-391). KX2-
391 was converted to the methanesulfonate salt by treatment with methanesulfonic acid
(MSA) in acetone at 50 ºC to afford 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-JV- benzylacetamide mesylate (KX2-391 MSA).
EXAMPLES Example 1: Small Scale Synthesis of KX2-391

[000343] The preliminary synthesis described below was illustrated in
US20060160800A1. This procedure is useful for small scale reactions, for example, reactions that produce up to 50 g of product.
[000344] For the following synthesis, unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton and carbon nuclear magnetic resonance spectra were obtained on a Bruker AC 300 or a Bruker AV 300 spectrometer at 300 MHz for proton and 75 MHz for carbon. Spectra are given in ppm (δ) and coupling constants, J, are reported in Hertz. Tetramethylsilane was used as an internal standard for proton spectra and the solvent peak was used as the reference peak for carbon spectra. Mass spectra and LC-MS mass data were obtained on a Perkin Elmer Sciex 100 atmospheric pressure ionization (APCI) mass spectrometer. LC-MS analyses were obtained using a Luna C8(2) Column (100 x 4.6 mm, Phenomenex) with UV detection at 254 nm using a standard solvent gradient program (Method B). Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by ultraviolet (UV) light, iodine, or 20 wt % phosphomolybdic acid in ethanol. HPLC analyses were obtained using a Prevail Cl 8 column (53 x 7 mm, Alltech) with UV detection at 254 nm using a standard solvent gradient program (Method A or B). Method A:
A = Water with 0.1 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.1 v/v Trifluoroacetic Acid

Method B:
A = Water with 0.02 v/v Trifluoroacetic Acid
B = Acetonitrile with 0.02 v/v Trifluoroacetic Acid

Synthesis of Η-benzyl-2- (5-bromopyridin-2-yl)acetamide :

[000345] A flask was charged with 5-(5-bromopyridin-2(lH)-ylidene)-2,2-dimethyl- l,3-dioxane-4,6-dione (1.039 g, 3.46 mmol), benzylamine (0.50 mL, 4.58 mmol), and toluene (20 mL). The reaction was brought to reflux under nitrogen for 18 hours, then cooled and placed in a freezer until cold. The product was collected by filtration and washed with hexanes to yield a mass of bright white crystals (1.018 g, 96%).
Synthesis of 4- (2- (4- (4, 4, 5, 5-tetramethylfl, 3, 2] dioxaborolan-2-yl)- phenoxy) ethyl)morpholine :

[000346] To a stirring solution of 4-(4,4,5,5-tetramethyl[l,3,2]dioxaborolan-2-yl)- phenol (2.55 g, 11.58 mmol), 2-morpholin-4-ylethanol (1.60 mL, 1.73 g, 13.2 mmol) and triphenyl phosphine (3.64 g, 13.9 mmol) in methylene chloride (60 mL) at 0 ºC was added dropwise DIAD (2.82 g, 13.9 mmol). The reaction was allowed to warm to room temperature and stir overnight. After 18 hours, additional portions of triphenyl phosphine (1.51 g, 5.8 mmol), 2-morpholin-4-ylethanol (0.70 mL, 5.8 mmol), and DIAD (1.17 g, 5.8 mmol) were added. After stirring an additional 2 hours at room temperature the reaction was concentrated and the residue purified by flash chromatography (5% to 25% EtOAc in CHCI3) to provide the product as a white solid (2.855 g, 74%).
Synthesis of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide KX2-391

[000347] A lO rnL reaction tube with a septum closure and stir bar was charged with N- benzyl-2-(5-bromopyridin-2-yl)acetamide (123 mg, 0.403 mmol), 4-(2-(4-(4,4,5,5- tetramethyl[l,3,2]dioxaborolan-2-yl)-phenoxy)ethyl)morpholine (171 mg, 0.513 mmol), and FibreCat 1007 (30 mg, 0.015 mmol). Ethanol (3 mL) was added, followed by aqueous potassium carbonate solution (0.60 mL, 1.0 M, 0.60 mmol). The tube was sealed and heated under microwave conditions at 150 ºC for 10 minutes. The reaction was cooled and concentrated to remove the majority of the ethanol, and then taken up in 10 mL of ethyl acetate and washed successively with water and saturated sodium chloride solution. The organic layer was dried with MgSO4, filtered and concentrated to a white solid. This white solid was triturated with ethyl ether to give KX2-391 as a white solid (137 mg, 79%): mp 135-137 ºC; 1H NMR (300 MHz,CDCl3) δ 8.70 (d, IH, J=2.0 Hz), 7.81 (dd, IH, J=2.4 Hz, J=8.0Hz), 7.65 (br s, IH), 7.49 (d, 2H, J=8.8 Hz), 7.37-7.20 (m, 6H), 7.01 (d, 2H, J=8.8 Hz), 4.49 (d, 2H, J=5.8 Hz), 4.16 (t, 2H, J=5.7 Hz, 3.82 (s, 2H), 3.78-3.72 (m, 4H), 2.84 (t, 2H, J=5.7 Hz), 2.62-2.58 (m, 4H); HPLC (Method B) 98.0% (AUC), tR = 1.834 min.; APCI MS m/z 432 [M+H]+.
Example 2: Intermediate Scale Synthesis of KX2-391 di-hydrochloride
[000348] The synthesis outlined in this example can be used on intermediate-scale reactions. The preparation of batches of at least 50 g of the dihydrochloride salt of KX2-391 is shown in Scheme 1. The linear synthesis consisted of 6 steps, a seventh step being the preparation of one of the reagents, 6-fluoropyridin-3-ylboronic acid (which is also available commercially). The overall yield of the sequence was 35% with an average yield of 83%, with the lowest yielding step being that of 68%. Of the seven steps only one required chromatography. The procedure listed below was performed on a 70 g scale.
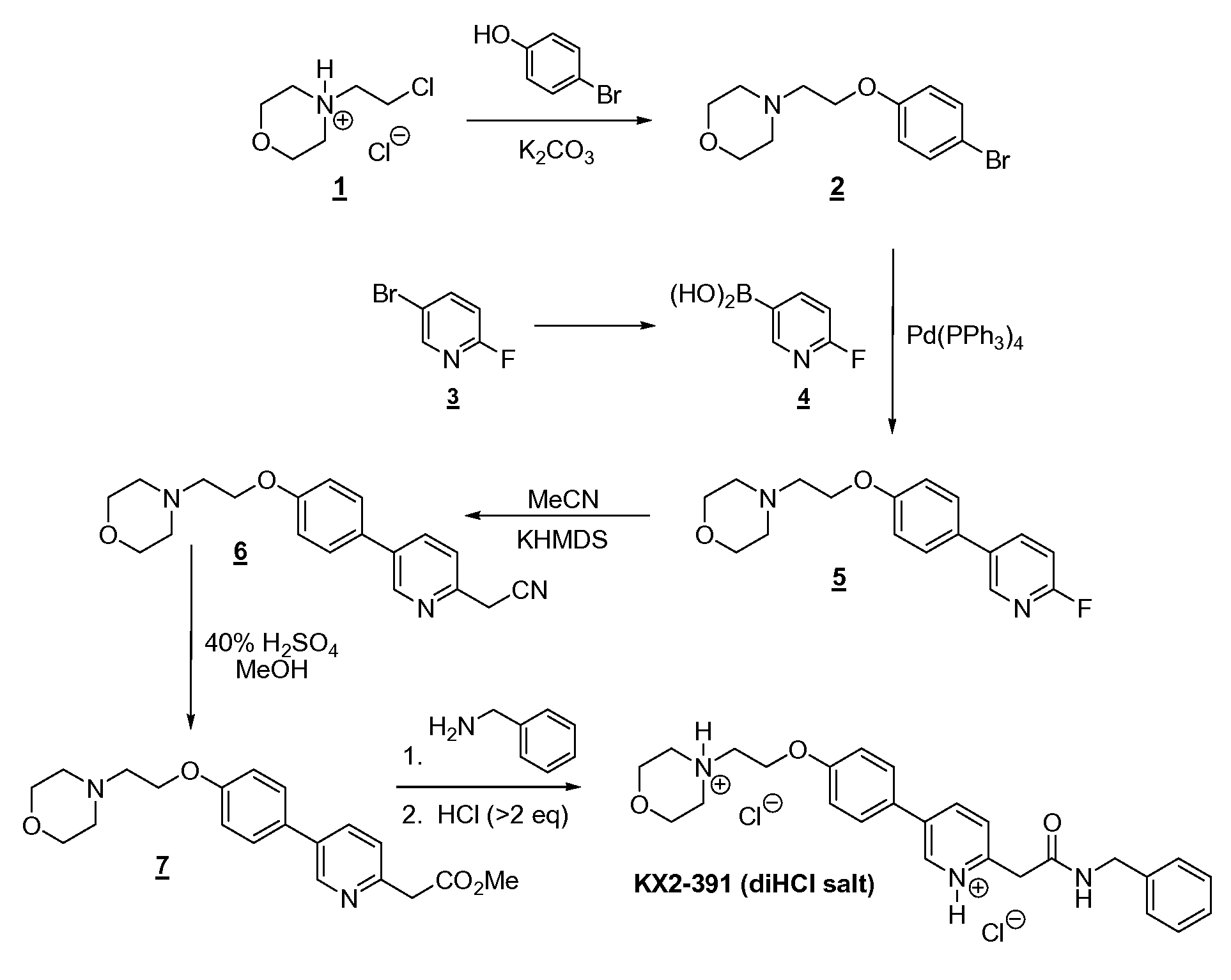
[000349] The first step is a Williamson ether synthesis between 4-bromophenol (131 g) and N-chloroethylmorpholine (1 as the HCl salt; 141 g) using K2CO3 powder (3 to 3.5 equivalents) as the base and having acetonitrile as the solvent. The ingredients were mixed and stirred at reflux overnight with high conversion (96.3-99.1%). After dilution with dichloromethane and heptane, the reaction mixture was filtered and evaporated to give the desired product 2 in essentially a quantitative yield (216 g). Note that with similar substrates (e.g., 4-bromo-3-fluorophenol), conversions (even with extensive heating) were not always so high (e.g., 59.9-98.3%). Both the alkyl chloride and the K2CO3 are preferably purchased from Aldrich. If continued heating does not drive reaction to completion, unreacted bromophenol can readily be removed by dissolving the crude reaction mixture in 4 parts toluene and washing out the phenol with 4 parts 15% aqueous NaOH. [000350] One of the reagents required for the second step (Suzuki coupling) was 6- fluoropyridin-3-ylboronic acid (4). Although available commercially, this reagent was readily prepared by lithium-bromide exchange of 5-bromo-2-fluoropyridine (3, 102 g) with n- butyllithium (1.2 eq) at low temperatures (<-60 ºC) in TBME followed by the addition of triisopropylborate (1.65 eq). Both stages of the reaction are brief, with an overall reaction time (including addition times) of ~3 h. Quenching is achieved with aqueous 24% NaOH, which also extracts the product leaving impurities in the organic layer. Once the aqueous layer is removed, it is then neutralized with HCl and extracted with EtOAc. After drying the organics and diluting with some heptane, concentration leads to precipitation/ crystallization of the product. Filtration gave the boronic acid 4 in relatively high purity (96.4% AUC) and good yield (69 g, 79-90%; see note on estimation of yield in the experimental section), which can be used without further purification.
[000351] The second reaction step in the linear sequence (a Suzuki coupling) is a simple reaction to set up; all the reagents [2 (111 g), aqueous Na2CO3, DME, and Pd(PPh3)4 (0.04 eq)] were charged to the reaction flask and the mixture heated at reflux; note that the reaction mixture was degassed to remove oxygen. Once the reaction is complete (within 7 h), the work-up involved decanting (or siphoning off) of reaction solution from the organic salts on the side of the flask (there was no visible aqueous layer), the flask was rinsed, and dried, and the solvent was removed from the combined organics. Crystallization of crude 5 from isopropanol/heptane provided material of improved purity compared to the crude, but still required chromatography (ratio of silica gel to crude was -8.5:1) to obtain material of adequate purity (>98%); the yield was 68% (79.5 g). Use of clean 5 prevented the need for chromatography in the next step, acetonitrile displacement of the fluorine atom. [000352] The replacement of fluoride with acetonitrile was also a simple reaction, and a simple room temperature crystallization of the crude product provided clean 6 in high yield and purity. The reaction involved initial formation of the “enolate” from acetonitrile (6.5 eq) using potassium hexamethyldisilane KHMDS (8 eq)/THF at -10 ºC followed immediately by the addition of fluoride 5 (79 g). The reaction was quick and after one hour quenching was achieved with saturated brine. After drying and evaporation of solvent of the organics, the resulting crude mixture consisted of only two components, the desired product and a much less polar product from apparent self-condensation of acetonitrile. The crude mixture was swirled in isopropanol/heptane and allowed to sit overnight, which resulted in complete crystallization of the product, which was filtered off and washed to provide high purity 6 (99.3% AUC) in good yield (64 g, 76%).
[000353] Methanolysis of 6 (64 g) was accomplished by heating in 40% H2SO4 (in
MeOH) until the reaction was complete (25 h). The reaction was then cooled, stirred with MgSO4 to convert traces of hydro lyzed product (ArCH2-CO2Me) back to product, and then added to cooled, aqueous K2CO3, with simultaneous extraction into dichloromethane. Drying and evaporation of most of the DCM followed by addition of 5% EtOAc (in heptane) and further concentration resulted in the crystallization of the product. Filtration of the solid and washing gave high purity (98.9% AUC) 7 in good yield (82%), additional high purity product (4 g) being obtained from the mother liquors for a total yield of 61.7 g (87%). [000354] The amidation step also involved charging of the reaction vessel with the ingredients (7 (61 g), benzyl amine (3 eq), and high boiling anisole) and then heating at reflux until the reaction was complete. Cooling of the reaction mixture resulted in complete crystallization of the target compound with high purity (98.9%) and good yield (81%). [000355] The final step was the formation of the dihydro chloric salt of the target compound. In order to ensure complete protonation at both basic sites, the reaction was conducted in absolute ethanol, which freely dissolved the dihydrochloride salt. After evaporation to near dryness, the reaction mixture was “chased” with ethanol twice to remove excess hydrogen chloride. The resulting viscous oil was dissolved in ethanol (2 parts) and then added, with rapid stirring, to a large volume (20 parts) EtOAc (ethyl acetate). Filtration, washing with ethyl acetate (no heptane) and vacuum drying provided the dihydrochloride salt of KX2-391 as a creamy-white powder. A total of 68 g (yield of 97%) was obtained of the final salt in high purity (99.6% AUC), which contained traces of EtOAc (4.8% w/w), EtOH (0.3% w/w), and heptane (0.6% w/w; from a final wash with heptane prior to vacuum drying). This salt was also crystallized (instead of the precipitation method described above) from hot EtOH/EtOAc to afford crystalline beads that had much lower entrapped solvent levels (only 0.26% w/w of EtOAc and 0.45% w/w of EtOH) and was free-flowing.

Preparation of 4-(2-(4-bromophenoxy)ethyl)morpholine (2):
[000356] A 5 L three-necked round-bottomed flask, equipped with mechanical stirrer, thermometer with adapter, condenser, and nitrogen inlet (on top of condenser), was charged with 1 (140.7 g, 0.756 mol), 4-bromophenol (130.6 g, 0.755 mol), anhydrous K2CO3 powder (367.6 g, 2.66 mol, 3.5 eq), and acetonitrile (1.3 L). The mixture was vigorously stirred (blade touching bottom of flask) at 80 ºC (overnight), followed by dilution with DCM (500 mL) and heptane (200 mL) and filtration through Celite. Evaporation to dryness (rotovap, then high vac) gave 2 as a light yellow oil (216.00 g, yield of 100%, 96.3% AUC, contains 3.7% unreacted bromophenol). This material was used successfully without further purification.
[000357] 1H NMR (CDCl3) δ 2.57 (t, 4 H), 2.79 (t, 2 H), 3.73 (t, 4 H), 4.08 (t, 2 H), 6.78
(d, 2 H), 7.37 (d, 2 H). MS (from LC/MS): m/z 287.1 [M + I].
[000358] That the bromophenol can be readily removed was demonstrated on a 2 g sample by first dissolving the sample in toluene (8 g) and washing with 8 g of 15% aqueous NaOH; liquid chromatography showed no trace of unreacted bromophenol in the recovered product (1.97 g; 98.5% recovery).

Preparation of 6-fluoropyridin-3-ylboronic acid (4):
[000359] To stirred and cooled (dry ice-acetone bath) anhydrous [TBME] (620 mL; in a
3 L three-necked round-bottomed flask equipped with mechanical stirrer, temperature probe with adapter, and nitrogen inlet) was added (via syringe) 2 M BuLi (352 mL, 0.704 mol, 1.2 eq). To this rapidly stirred and cooled (< -75 ºC) mixture was added a solution of 3 (102.2 g, 0.581 mol) in anhydrous TBME (100 mL) over a period of 13 min during which time the internal temperature rose to -62 ºC. The reaction was stirred for another 45 min (the temperature was maintained between -62 ºC and -80 ºC), followed by the rapid and sequential addition of four portions of triisopropylborate (total of 180 g, 0.957 mol, 1.65 eq). At the end of the addition the internal temperature had risen to -33 ºC. After stirring an additional 45 min over the cold bath (internal temperature lowered from -33 ºC to -65 ºC), the cold bath was removed and the stirred mixture on its own rose to -22 ºC over a period of 50 min. After warming (via water bath) to 6 ºC over a period of 15 min, the stirred reaction mixture was placed in an ice-water bath and then quenched under nitrogen with a cooled solution of NaOH (160 g) in water (500 mL). Once the addition was complete, the internal temperature was 20 ºC. This mixture was stirred at room temperature for 1.5 h. The aqueous layer was removed, neutralized to pH 7 with -350 mL concentrated HCl, and then extracted with EtOAc (3 x 1 L). Because the pH was now 8-9, the aqueous layer was adjusted to pH 7 using ~15 mL concentrated HCl and extracted further (2 x 1 L) with ethyl acetate. The combined EtOAc extracts were dried (Na2SO4), filtered, and concentrated to a volume of -150 mL. With swirling of the concentrate, heptane was added in portions (total volume of 300 mL) resulting in the precipitation/crystallization of the product. Filtration, washing of the solid with heptane (100 mL, 300 mL, then another 300 mL), and air drying gave the title product as an off-white solid (68.6 g, yield of 79-90%*; LC purity of 96.4%, NMR showed an estimated 5.5% w/w of heptane), which was used successfully without further purification. LC/MS showed it to be a mixture of the two following entities, the intensity of the higher molecular weight entity being major (*Note: yield of reaction is 79% if the boronic acid is assumed to be the only constituent and is 90% if it is assumed that the cyclic borate is the only constituent):

1H NMR (CDCl3) δ 7.14 (dd, 1 H), 8.27 (ddd, 1 H), 8.39 (br s, 2 H, 2 OH), 8.54 (fine d, 1 H). MS (from LC/MS): m/z 143.0 [M + 1; for boronic acid] and 370.0 [M + 1; for cyclic borate above].

[000360] A 2 L three-necked round-bottomed flask equipped with mechanical stirrer, thermometer and adapter, condenser, and nitrogen inlet (at top of condenser) was charged with 2 (110.7 g, 0.387 mol), 4 (71.05 g, 0.477 mol, 1.23 eq) and DME (700 mL). The resulting stirred solution was degassed by passing a rapid stream of nitrogen through the stirred solution over a period of 5 min followed by the addition of a degassed solution of Na2CO3 (121.06 g, 1.142 mol, 3 eq) in H2O (250 mL) and also solid Pd(PPh3)4 (19.8 g, 0.044 eq). Immediately after the last addition, the head space above the reaction mixture was purged with nitrogen and the mixture then stirred at 80-85 ºC (internal temperature) for 7 h, followed by cooling to room temperature. Because of the lack of an aqueous layer, the supernatant was decanted, leaving behind the inorganic salts (with adsorbed water). The reaction flask with the inorganic salts was washed with 50% dichloromethane/ethyl acetate (2 x 250 mL), the washes being added to the decanted supernatant. These combined organics were dried (Na2SO4), filtered, and evaporated to dryness to a dark brown oil (148 g). To this oil was added 15O g of 50% heptane/isopropyl alcohol (IPA) and after swirling and cooling (via ice water bath), crystallization began. Additional heptane (50 g) was added and the resulting solid was filtered, washed, and air dried to give 48 g of a light brown solid. After evaporating the filtrate to dryness, the resulting mixture was swirled in 100 mL of 50% heptane/IPA followed by the addition of more heptane (-100 mL), stoppering and placing in the freezer for crystallization. The resulting solid was filtered, washed with heptane, and air dried to give 61 g of a gummy solid. Evaporation of the resulting filtrate gave an oil (34 g) which contained significant less polar impurities including Ph3P=O and so it was partitioned between 2 N HCl (240 mL) and EtOAc (220 mL). The bottom aqueous layer was removed and then stirred with EtOAc while neutralizing with K2CO3 to a pH of 7-8. The EtOAc layer was dried, filtered, and evaporated to dryness (22 g). The 48 g, 61 g, and 22 g portions were chromato graphed over silica gel (1.1 Kg) packed in DCM. Elution with DCM (400 mL), 50% DCM/EtOAc (5 L), and then 50% DCM/EtOAc (8 L) containing increasing amounts of MeOH/Et3N (beginning with 1.5% MeOH/1% Et3N and ending with 5% MeOH/3% Et3N) gave 77.68 g of a viscous oil (purity 98.0%) which immediately crystallized upon swirling in heptane (300 mL). Filtration, washing with heptane and air drying gave 75.55 g (98.7% AUC) of solid 5. Additional pure 5 (total of 3.9 g, 98.6-99.3% AUC) was obtained from earlier chromatographic fractions containing Ph3P=O by cleaning them up as done for the above 34 g sample, followed by evaporative crystallization. The total yield of 5 was 79.5 g (68%). 1H NMR (CDCl3) δ 2.59 (t, 4 H), 2.84 (t, 2 H), 3.75 (t, 4 H), 4.16 (t, 2 H), 6.97 (dd, 1 H), 7.01 (d, 2 H), 7.46 (d, 2 H), 7.92 (ddd, 1 H), 8.37 (fine d, 1 H). MS (from LC/MS): m/z 303.2 [M + I].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6):
[000361] A 3 L three-necked round-bottomed flask was equipped with mechanical stirrer, thermometer and adapter, additional funnel, and nitrogen inlet (on top of addition funnel, positive pressure through a bubbler). With a rapid stream of nitrogen going through the bubbler, the stopper was removed and the flask was charged with KHMDS (415.8 g, 2.08 mol) and then anhydrous THF (1 L). To the stirred and cooled (ice/methanol bath, internal temperature of solution was -8 ºC) KHMDS/THF solution was added dropwise a solution of MeCN (70 g) in THF (110 mL) over a period of 22 min followed immediately by the relatively rapid (4 min) addition of a solution of 5 (79.06 g, 0.262 mol) in THF (400 mL), after which time the internal temperature of the reaction mixture had reached 10 ºC. With continued cooling (1 h) the internal temperature was -6 ºC and by TLC the reaction appeared complete. After an additional 30 min (internal temperature of -3 ºC), the reaction mixture was quenched with saturated brine (1 L) and diluted with EtOAc (500 mL). After removing the aqueous layer, the organic solution was dried (Na2SO4), filtered, and evaporated to dryness (to an oil) followed by completely dissolving in IPA (150 mL), diluting with heptane (300 mL), adding seed crystals (prepared by dissolving -100 mg of crude oil in IPA (-150 mg) and diluting with heptane (-2.5 mL)), and allowing to stand overnight. After stirring to break up the crystalline solid, the solid was filtered, washed with 250 mL 2:1 heptane/IP A and then multiple washes with heptane and air dried to give 64.38 g (yield of 76%) of title product 6 as a crystalline tan solid (LC purity of 99.3%). Another 5.88 g of less pure material was obtained from the filtrate.
[000362] 1H NMR (CDCl3) δ 2.59 (t, 4 H), 2.84 (t, 2 H), 3.74 (t, 4 H), 3.97 (s, 2 H),
4.17 (t, 2 H), 7.02 (d, 2 H), 7.46 (d, 1 H), 7.51 (d, 2 H), 7.87 (dd, 1 H), 8.77 (fine d, 1 H). MS (from LC/MS): m/z 324 A [M + I].

Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7): [000363] A 2 L single-necked round-bottomed flask was charged with 6 (64.00 g, 0.198 mol) and MeOH (360 g) followed by the slow, careful, and dropwise addition OfH2SO4 (240 g) and the resulting homogeneous solution stirred at reflux (115 ºC oil bath) until the reaction was complete (25 h with 0.8% unreacted starting material) with 3.5% ArCH2CO2H. After brief cooling, MgSO4 (75 g) was added and the mixture swirled and allowed to stand an additional 45 min (composition now 96.3% product, 0.8% unreacted starting material, and 2.5% ArCH2CO2H). The reaction mixture was then added slowly to a rapidly stirred and cooled (ice-water bath) mixture of DCM (2 L) and a solution OfK2CO3 (450 g) in H2O (600 mL). The resulting emulsion was allowed to stand overnight. The clear portions of organic solution were siphoned off and the remainder portions were treated iteratively with water and DCM, the clear organics being combined with the original portion that was siphoned off. The combined organics were dried (Na2SO4), filtered, and concentrated to a volume of ~1.2 L followed by the addition of 300 mL of 5% EtOAc (in heptane) and then heptane (300 mL) and the mixture concentrated (rotovap with heat) again to remove the DCM. At this point 15 mL EtOAc was added and the hot mixture swirled until crystallization had begun, swirling continued until crystallization was near complete, and then allowed to stand and cool to room temperature for complete crystallization. The solid was then filtered, washed with 300 mL 5% EtOAc (in heptane) and heptane (100 mL) and then fully air dried to give 57.74 g (yield of 82%) of 7 as a light yellow solid (98.9% AUC). Another 3.94 g of clean product (97.9% AUC) was obtained from the filtrate (total yield of 87%).
[000364] 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.84 (t, 2 H), 3.74 (overlapping t and s, 6 H),
3.89 (s, 2 H), 4.17 (t, 2 H), 7.01 (d, 2 H), 7.34 (d, 1 H), 7.49 (d, 2 H), 7.80 (dd, 1 H), 8.74 (fine d, 1 H). MS (from LC/MS): m/z 357.4 [M + I].

Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391 free base).
[000365] A l L single-necked round-bottomed flask was charged with 7 (61.4 g, 0.172 mol), benzyl amine (55.6 g, 0.519 mol, 3 eq), and anhydrous anisole (300 g) and then stirred at reflux until reaction was essentially complete (23 h, 165 ºC oil bath temperature; internal temperature was 147 ºC) and then allowed to cool to near room temperature. A portion (1 mL) of the reaction mixture was diluted with toluene (1 mL) resulting in the complete crystallization of that portion. This seed was then added to the reaction mixture and allowed to stand until the whole reaction mixture had crystallized to a single block. Toluene (150 mL) was added and the mixture swirled to break up the solid. Heptane/toluene (1 :1, 100 mL) was added and the solid mixture broken up further. Finally, heptane (50 mL, then 25 mL) was added and the mixture broken up even further, allowing to stand an additional 30 min before filtering the solid. Filtration of the solid, washing with 2:1 toluene/heptane (300 mL), 1 :2 toluene/heptane (300 mL), and then heptane (2 x 300 mL), and then drying (air, then high vac) gave 60.16 g (yield of 81%) of title product as a white solid (≥98.9% AUC). Another 2.5 g of less pure (97.4%) material was obtained from the mother liquors. 1H NMR (CDCl3) δ 2.60 (t, 4 H), 2.83 (t, 2 H), 3.74 (t, 4 H), 3.82 (s, 2 H), 4.18 (t, 2 H), 4.49 (d, 2 H), 7.01 (d, 2 H), 7.2-7.35 (m, 6 H), 7.49 (d, 2 H), 7.64 (br t, 1 H), 7.81 (dd, 1 H), 8.69 (fine d, 1 H). MS (from LC/MS): m/z 432.5 [M + I].
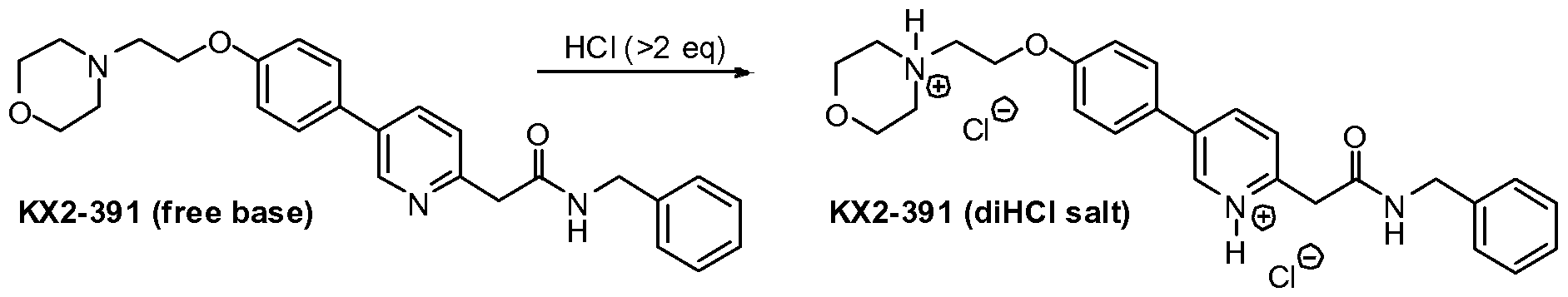
Preparation of 4-(2-(4-(6-(2-(benzylamino)-2-oxoethyl)pyridinium-3-yl)phenoxy)ethyl)- morpholin-4-ium chloride (KX2-391, diHCl salt).
[000366] To a stirred suspension of KX2-391 (free base, 60.00 g) in absolute EtOH (600 niL) was added 170 niL of 2.5 M HCl (in ethanol), 25 niL EtOH being added to wash down the sides of the flask. The resulting homogeneous solution was stirred at room temperature (20 min) and then evaporated to near dryness (to frothing). After chasing with EtOH (2 x 150 mL), the residue was taken up again in EtOH (150 mL) and then was followed by the slow addition of heptane until the mixture appeared saturated (33 mL required for cloudiness to remain). After sitting overnight, two layers had formed. After adding additional heptane (250 mL) crystallization still could not be induced and so the reaction mixture was concentrated to a volume of -200 mL at which time the mixture was homogeneous. This thick homogeneous solution was added dropwise to very rapidly stirred (mechanical) EtOAc (2 L). After the addition was complete, a 25 mL EtOH rinse of the original flask and addition funnel was added to the rapidly stirred mixture. The rapid stirring was continued for another ~1 h and then the mixture was filtered and the solid (partly gummy) was washed with EtOAc (300 mL) and then heptane. As soon as the heptane wash began, the solid got much gummier. The fritted Buchner funnel and its contents were covered (paper towel/rubber band) and immediately placed in the vacuum oven. After overnight vacuum at ~45 ºC, the vacuum was released under nitrogen, and the Buchner funnel containing the product (foamy solid) was immediately placed in a zip-lock back and then, under nitrogen (glove bag), transferred to a bottle and the foamy solid broken up (spatula) to a powder. A second night under high vacuum (-45 ºC) resulted in only 1.3 g of additional weight loss. Constant weight was essentially attained with the third night of high vacuum (~45 ºC) where only 0.2 g of weight was lost. The final weight of material was 68.05 g (yield of 97%), containing 0.29 eq (4.8% w/w) of EtOAc, 0.035 eq (0.3% w/w) EtOH, and 0.03 eq (0.6% w/w) heptane. The purity was 99.6%.
1H NMR (DMSO-d6) δ 3.1-3.3 (m, 2 H), 3.45-3.65 (m, 4 H), 3.8-4.0 (m, 4 H), 4.11 (s, 2 H), 4.32 (d, 2 H), 4.57 (t, 2 H), 7.19 (d, 2 H), 7.2-7.4 (m, 5 H), 7.88 (d, 2 H), 7.93 (d, 1 H), 8.68 (dd, 1 H), 8.99 (br t, 1 H), 9.10 (fine d, 1 H), 11.8 (br s, 1 H). MS (from LC/MS): m/z 432.5 [M + 1 of free base].
Elemental analysis (for C26H29N3O3 • 2 HCl • 0.035 EtOH • 0.29 EtOAc • 0.03 heptane • 0.8 H2O): Calculated (%): C, 60.03; H, 6.54; N, 7.65; Cl, 12.91 Observed (%):C, 59.85/59.97; H, 6.54/6.47; N, 7.67/7.67; Cl, 13.10/13.24 Calculated FW: 534.63 (does not take into account the 0.8 H2O which probably arose during handling of this very hygroscopic powder, since 1H NMR shows no evidence for H2O). [000367] The ethyl chloride level in this material was measured and found to be 98 ppm. The sample was also analyzed and found to contain 5,800 ppm of heptane. [000368] Analysis of another portion of this sample yielded the following results: 99.6% AUC, 1640 ppm ethanol, 41,480 ppm ethyl acetate, 5600 ppm heptane, no anisole detected, and 120 ppm ethyl chloride.
[000369] A procedure for recrystallizing the salt was also developed using the above dried salt. This procedure would work just was well on the highly pure crude salt (containing residual EtOH) obtained from concentrating the HCl salt-forming reaction mixture: [000370] The salt (575 mg) was dissolved in twice the mass of absolute EtOH (1.157 g) and then heated under nitrogen. To this hot solution (stirred) was added 1.6 g of 25% EtOH (in EtOAc) followed by the addition of EtOAc (0.25 mL) resulting in a cloudiness that remained. The cloudy hot solution was allowed to cool to room temperature during which time crystallization occurred. After crystallization was complete (2 h), the crystalline solid was filtered, washed with anhydrous EtOAc (~40 mL), and vacuum dried to give 424 mg of the dihydrochloride salt of KX2-391 as a free-flowing solid (tiny beads, 99.8% AUC) containing only 0.05 eq (0.45% w/w) of EtOH and 0.015 eq (0.26% w/w) of EtOAc. Slightly better recovery (460 mg from 586 mg) was attained using isopropanol/EtOAc but the level of solvent entrapment was higher [0.085 eq (1.0% w/w) of isopropanol and 0.023 eq (0.4% w/w) OfEtOAc].
Example 3: Large Scale Synthesis of KX2-391 di-HCl
[000371] Reagents and solvents were used as received from commercial suppliers.
Progress of the reactions was monitored by HPLC, GC/MS, or 1H NMR. Thin-layer chromatography (TLC) was performed using Analtech silica gel plates and visualized by UV light (254 nm). High pressure liquid chromatography (HPLC) was performed on an Agilent 1100 Series instruments. Proton and carbon nuclear magnetic resonance spectra were obtained using a Bruker AV 300 at 300 MHz for proton and 75 MHz for carbon. The solvent peak was used as the reference peak for proton and carbon spectra.
Preparation of 4-(2-(4-Bromophenoxy)ethyl)morpholine (2) [000372] A 50 L jacketed reactor equipped with a reflux condenser and temperature probe was charged with 4-(3-chloropropyl)morpholine (2.44 kg, 0.54 mol), 4-bromophenol (2.27 kg, 0.54 mol, 1.0 equiv.), powdered potassium carbonate (6.331 kg, 1.88 mol, 3.50 equiv.), and DMF (12.2 L) and stirred. The reaction mixture was then heated to 60-65 ºC and stirred overnight. After 17.5 h, the reaction mixture was cooled to 20-25 ºC. The reaction mixture was charged to a different reactor equipped with bottom valve for the work-up. While maintaining a temperature between 20-30 ºC, DI water (48.7 L) was charged to the reactor. The phases were separated. The aqueous layer was extracted with MTBE (3 x 24.4 L). To the combined organics, DI water (18.3 L) and then 6M sodium hydroxide (18.2 L) were added. The mixture was stirred for 2-5 minutes and the phases were separated. The organic phase was washed with water (24.4 L) and brine (24.4 L), dried over magnesium sulfate, filtered, and concentrated to give 337Og of a yellow oil (89% crude yield, 99.4% AUC by HPLC).
Preparation of 6-fluoropyridin-3-ylboronic acid (4)
[000373] A 72 L reactor equipped with reflux condenser, and temperature probe. To the reactor 5-bromo-2-fluoropyridine (1.17 L, 0.568 mol), toluene (18.2 L), and triisopropyl borate (3.13 L, 0.68 mol, 1.2 equiv.) were charged and stirred. Tetrahydrofuran (4.4 L) was added to the reactor and the reaction mixture was cooled to between -35 to -50 ºC. While maintaining a temperature between -35 to -45 ºC, n-butyl lithium (2.5 M solution of hexanes, 5.44 L, 0.68 mol, 1.2 equiv.) was cautiously added to the reactor. After 5 h, the reaction was deemed complete and the reaction mixture was warmed to between -15 to -20 ºC. To the reaction was added 2M HCl (11.80L) to the reactor while maintaining a temperature between -15 ºC and 0 ºC. The reaction mixture was stirred at 18 to 23 ºC for (16 h) and the phases were separated. The organics were then extracted with 6 M sodium hydroxide (6.0 L). The acidic anbasic aqueous phases were mixed in the reactor and 6 M HCl (2.5 L) was added until pH 7.5 was achieved. Sodium chloride (6.0 kg) was then added to the aqueous phase. The aqueous phase was then extracted with THF (3 x 20 L). The combined organics were dried with magnesium sulfate and concentrated to give 1300 g of a tan solid (81% crude yield).
Preparation of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000374] A 72 L reactor equipped with reflux condenser, sparging tube, bubbler, and temperature probe was charged with 6-fluoropyridin-3-ylboric acid (2.84 kg, 1.24 equiv.), A- (2-(4-bromophenoxy)ethyl)morpholine (4.27 kg, 1.0 equiv.), and DME (27 L). Agitation was started and sodium carbonate (4.74 kg, 3.0 equiv.) as a solution in DI water (17.1 L) was then charged to the reaction mixture. Argon was bubbled through the reaction mixture for 50 minutes. Under an argon atmosphere, tetrakis(triphenylphosphine)palladium (750 g, 0.04 equiv.) was added to the reaction mixture as a slurry in DME (1.0 L). The reaction mixture was heated to 75 – 85 ºC and stirred overnight (17 h). The reaction mixture was cooled to between 18 – 22ºC. DI water (26.681kg) and MTBE (26.681 L) were charged to the reactor and stirred for 5 minutes. The phases were separated and the aqueous phase was extracted with MTBE (2 x 26.7 L). The combined organics were extracted with 2M HCl (1 x 15.0 L, 3 x 21.8 L). The aqueous phase was then charged back to the reactor and ethyl acetate was added (26.7 L). The pH was adjusted to 6.2 using 6 M sodium hydroxide (26.7 L) while maintaining a temperature between 15 – 25 ºC. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 x 26.7 L). The combined organics were dried with magnesium sulfate and concentrated to give 4555 g of a residue (101% crude yield, 67.1% AUC by HPLC).
Purification of 4-(2-(4-(6-fluoropyridin-3-yl)phenoxy)ethyl)morpholine (5)
[000375] The crude product (575 g) was purified by silica gel chromatography by eluting with methanol/ethyl acetate/heptane (30% ethyl acetate/heptane, 50% ethyl acetate/heptane, 75% ethyl acetate/heptane, 100% ethyl acetate, and 5% methanol/ethyl acetate). Concentration of the pure fractions by TLC (10% methanol/dichloromethane, Rf = 0.3) provided 420 g of a light brown solid (73% recovery, >99.9% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetonitrile (6)
[000376] A 1 M solution of NaHMDS (2.0 L, 5.0 equiv.) in THF was charged to a 5-L flask and cooled to -20 to -15 ºC. While maintaining a temperature below -10 ºC, fluoride (119.7g, 1.0 equiv.) in THF (500 mL) was charged to the flask over 20 minutes. Acetonitrile (82.5 mL, 4.0 equiv.) in THF (170 mL) was added to the flask over 20 minutes, while maintaining a temperature below -10 ºC. The reaction mixture was then stirred for 1 h. To the reaction was added brine (1.5 L, 12.6 vol.) at a rate as to maintain a temperature below 10 ºC. The solution was then warmed to room temperature and the layers were allowed to separate. The mixture was filtered over Celite and washed with THF (1 x 200 mL, 1 x 100 mL). The aqueous phase was extracted with toluene (750 mL). The combined organics were dried with magnesium sulfate, filtered, washed with toluene (2 x 25OmL), and concentrated to dryness. Toluene (IL) was added and the solution was concentrated to dryness again to give 169.8 g of an oil. MTBE (1190 niL, 7 vol.) was added to the oil at 50 ºC and stirred for 15 minutes. Heptane (850 rnL, 5vol.) was added over ten minutes at 50 ºC. The mixture was then cooled to room temperature over 1.5 h and stirred for 2 h. The slurry was filtered, washed with 1 :4 MBTE/heptane (2 x 100 mL), and dried in an oven overnight at 45 ºC to give 102.3 g of an off-white solid (80% yield, 98.8% AUC by HPLC).
Preparation of methyl 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)acetate (7)
[000377] Nitrile 6 (101 g) and methanol (1.01 L, 10 vol.) were charged to a 3-L flask equipped with stir bar and thermocouple. Concentrated H2SO4 (175 mL, 10.0 equiv.) was added drop wise to the solution over 15 minutes while maintaining a temperature below 60 ºC. Followed by 30% fuming sulfuric acid (124 mL) was added drop wise to the solution while maintaining a temperature below 60 ºC. The solution was then heated to reflux with a heating mantle and stirred overnight. When the reaction was deemed complete, it was cooled to 20 ºC. In a second flask (22 L), saturated sodium bicarbonate (10.7 L) and dichloromethane (1.1 L) were charged and cooled to 15 ºC. While maintaining a temperature below 20 ºC, the reaction mixture was added to the sodium bicarbonate/dichloromethane mixture. The quench was stirred for 15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (I x 55OmL, 1 x 30OmL). The combined organics were dried with magnesium sulfate and concentrated to dryness to give 105 g of an orange solid (94% crude yield, 97.7% AUC by HPLC).
Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide (KX2-391)
[000378] Ester 7 (103 g), anisole (513 mL, 5 vol.), and benzylamine (94 mL, 3.0 equiv.) were charged to a 3 L flask equipped with thermocouple and overhead stirrer. The reaction mixture was then heated to 142 ºC and stirred for two days. The reaction mixture was cooled to 45-50 ºC and stirred for 2 hours. To the mixture was added n-heptane (1.5 L) dropwise over an hour. The solution was cooled to room temperature over three hours and then stirred overnight. The resulting slurry was filtered, washed with 4:1 Anisole/n-heptane (200 mL) and n-heptane (3 χ100 mL). Drying in the oven overnight, the resulting product was 112. Ig of a tan solid (90% yield, 99.6% AUC by HPLC). The use of a single isomer of heptane was essential to adequately quantitate the residual solvent. See Figure 5 for 1H NMR of KX2- 391. Preparation of 2-(5-(4-(2-morpholinoethoxy)phenyl)pyridin-2-yl)-N-benzylacetamide dihydrochloride salt (KX2-391 2HC1)
[000379] EtOH (1.0 L) was charged to a 2-L flask and acetyl chloride (62.5 niL, 3.0 equiv.) was added slowly to the flask and stirred for 40 minutes. The resulting solution was added to KX2-391 (100 g) over 30 minutes while maintaining a temperature of 30 ºC. The solution was concentrated to a mass of 270 g. The concentrated solution was added to ethyl acetate (2 L) over 20 minutes with rapid stirring. The mixture was stirred overnight and then filtered under nitrogen to give two distinct solid products, tan solids (73.5 g) and darker solids (42.2 g). The solids were dry blended to give a combined yield of 99%. The HPLC analysis indicated 99.0% purity (AUC). Analysis indicated that ethanol was present at 2530 ppm, ethyl acetate at 48,110 ppm, ethyl chloride at 170 ppm, and no heptane and anisole were detected. Palladium content was assayed three times and measured to be 29 ppm, 2 ppm, and less than 1 ppm.
PATENT
CN 106810490
| 2-(5-(4-(2-morpholinylethoxy)phenyl)pyridine-2-yl)-N-benzyl-acetamide, development code KX -01, KX2-391, have the structure shown in formula I. |
| |
| Patent CN10118473B and US7300931B disclose compound KX2-391, and disclose its application in the treatment of cell proliferative disorders. KX2-391 and its pharmaceutically acceptable salts are effective Src tyrosine kinase inhibitors, which can effectively treat diseases and disorders regulated by Src kinase. KX2-391 has a GI50 of 9-60 nM in cancer cell lines and is currently in clinical phase II. |
| KX2-391 has polymorphism. Polymorphism refers to the phenomenon that the same compound can form two or more molecular spatial arrangements by controlling its different production conditions to produce different solid crystals. Different crystal forms of the same compound have the same chemical composition. , But the microscopic crystal structure is different, which leads to differences in their appearance, physical and chemical properties and biological activity. The phenomenon of polymorphism directly affects the processing performance of the drug formulation, and affects the stability, solubility, and bioavailability of the drug, and further affects the quality, safety, effectiveness and application of the drug. Therefore, in drug research and development, the polymorphism of drugs should be fully considered. At present, KX2-391 is still in the research and development stage, and a comprehensive study of its solid form is of great significance to the research and development of KX2-391 and the approval of the market. |
| Example 1 |
| 2-(5-(4-(2-morpholinylethoxy)phenyl)pyridin-2-yl)-N-benzyl-acetamide (KX2-391) crystal form (i.e. having formula (I) The structure of the crystalline diaryl compound, the subsequent examples are referred to as the preparation of KX2-391 crystal form B) |
| Put KX2-391 (5.0g) in a 500ml round bottom flask, add 150ml methanol to dissolve KX2-391 completely, and place it at 50°C and stir. 300ml of purified water was gradually added dropwise. After the addition, the resulting slurry was stirred at room temperature for 1 hour to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. The purity detected by HPLC is ≥99.83%. |
| Example 2 Preparation of KX2-391 crystal form B |
| Put KX2-391 (5.0g) in a 100ml round bottom flask, add 25ml of DMSO to dissolve KX2-391 completely, and stir at room temperature. Gradually add 50ml of purified water dropwise. After the dropwise addition, the resulting slurry was stirred at 0°C for 1h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%. |
| Example 3 Preparation of KX2-391 crystal form B |
| Put KX2-391 (5.0g) in a 250ml round bottom flask, add 15ml of dichloromethane to dissolve KX2-391 completely, and stir at 30°C. Gradually add 100ml of n-heptane dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 0.5h to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.80%. |
| Example 4 Preparation of KX2-391 crystal form B |
| Put KX2-391 (2.0g) in a 500ml round bottom flask, add 100ml of acetone to completely dissolve KX2-391, and stir at room temperature. Gradually add 150 ml of n-hexane, and after the addition is complete, the resulting slurry is stirred at 0°C for 1 h to crystallize, filtered with suction, and dried in vacuum at 50°C. The obtained solid is KX2-391 crystal form B. HPLC detection purity ≥99.79%. |
| Example 5 Preparation of KX2-391 crystal form B |
| Put KX2-391 (2.0g) in a 250ml round bottom flask, add 50ml of THF to dissolve KX2-391 completely, and place it at 40°C and stir. Gradually add 100 ml of methyl tert-butyl ether dropwise. After the dropwise addition, the resulting slurry was stirred at room temperature for 2 hours to crystallize, filtered with suction, and dried under vacuum at 50°C. The resulting solid was KX2-391 crystal form B. HPLC detection purity ≥99.81%. |
| Example 6 Detection of KX2-391 crystal form B |
| The KX2-391 crystal form B prepared in Example 1 was tested by XRPD method. The equipment used is RIGAKU TTR III X-ray powder diffractometer, measurement conditions and methods: Cu (target), 40KV-30mA (working voltage and current), 2θ=2~50 degrees (scanning range), 4.0deg /min. (scanning speed), the obtained spectrum is shown in Figure 1. It can be seen from Figure 1 that the XRPD spectrum of KX2-391 crystal form B provided in Example 1 is 2.10, 3.68, 4.16, 6.24, 8.33, There are peaks at 12.53, 16.26, 16.75, 18.33, 19.05, 19.85, 21.00, 21.50, 21.92, 22.50, 23.16, 25.08, 25.35, 25.70, 27.49, 29.67, 33.97, and 38.43. |
| The invention also adopts the DSC-TGA method to detect the crystal form B of KX2-391 provided by the invention. The equipment used is METTLER TOLEDO’s TGA-DSC, testing environment conditions 22℃, relative humidity RH68%, temperature range 0-400℃, heating rate 12℃/min, protective gas N 2 , The resulting maps are shown in Figure 2 and Figure 3. It can be seen from Figure 2 that the DSC spectrum of KX2-391 crystal form B provided in Example 1 has endothermic peaks at 126.9°C and 137.4°C. It can be seen from Figure 3 that the TGA pattern of KX2-391 crystal form B provided in Example 1 has no significant weight loss before 200°C. |
PAPER
Journal of Medicinal Chemistry (2018), 61(11), 4704-4719.
[1]. Lau GM, et al. Expression of Src and FAK in hepatocellul
https://pubs.acs.org/doi/10.1021/acs.jmedchem.8b00164
Abstract

The discovery of potent, peptide site directed, tyrosine kinase inhibitors has remained an elusive goal. Herein we describe the discovery of two such clinical candidates that inhibit the tyrosine kinase Src. Compound 1 is a phase 3 clinical trial candidate that is likely to provide a first in class topical treatment for actinic keratosis (AK) with good efficacy and dramatically less toxicity compared to existing standard therapy. Compound 2 is a phase 1 clinical trial candidate that is likely to provide a first in class treatment of malignant glioblastoma and induces 30% long-term complete tumor remission in animal models. The discovery strategy for these compounds iteratively utilized molecular modeling, along with the synthesis and testing of increasingly elaborated proof of concept compounds, until the final clinical candidates were arrived at. This was followed with mechanism of action (MOA) studies that revealed tubulin polymerization inhibition as the second MOA.

[1]. Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig Dis Sci, 2009, 54(7), 1465-1474.
//////////Tirbanibulin, Klisyri, FDA 2020, 2020 APPROVALS, KX2 391, KX 2391, KX-01, actinic Keratosis
O=C(CC1=NC=C(C2=CC=C(OCCN3CCOCC3)C=C2)C=C1)NCC4=CC=CC=C4