
DESIDUSTAT
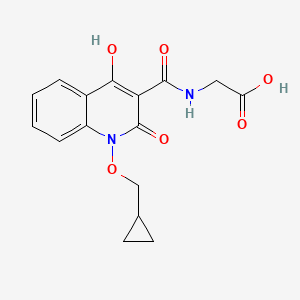
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%

Desidustat (INN, also known as ZYAN1) is an investigational drug for the treatment of anemia of chronic kidney disease. Clinical trials on desidustat have been done in India and Australia.[1] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean Hb increase of 1.57, 2.22, and 2.92 g/dL in Desidustat 100, 150, and 200 mg arms, respectively, was observed.[2] It is currently undergoing Phase 3 clinical trials.[3] Desidustat is being developed for the treatment of anemia, where currently erythropoietin and its analogues are drugs of choice. Desidustat is a prolyl hydroxylase domain (PHD) inhibitor. In preclinical studies, effect of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[4][5] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[6]. In January 2020, Zydus entered into licensing agreement with China Medical System Holdings for development and commercialization of Desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of Desidustat in Greater China [7]
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC5766731. PMID28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID30458168. S2CID53943666.
- ^ “Zydus enters into licensing agreement with China Medical System Holdings”. 20 January 2020. Retrieved 20 January 2020 – via Business Standard.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
|
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1