
Rimegepant
- Molecular FormulaC28H28F2N6O3
- Monoisotopic mass534.219116 Da
Antimigraine, Calcitonin receptor-like receptor antagonist
Treatment of migraine


|
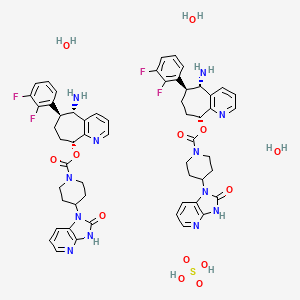
Rimegepant sulfate (USAN)
リメゲパント硫酸塩; |
|
| Formula |
(C28H28F2N6O3)2. H2SO4. 3H2O
|
|---|---|
| CAS |
1374024-48-2
|
| Mol weight |
1221.2386
|
Nurtec ODT, FDA 2020, 2020/2/27 fda approved
Bristol-Myers Squibb developed Rimegepant, also known as BMS-927711. Rimegepant is a potent, selective, competitive and orally active calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. Rimegepant has shown in vivo efficacy without vasoconstrictor effect; it is superior to placebo at several different doses (75 mg, 150 mg, and 300 mg) and has an excellent tolerability profile.
Rimegepant is a medication for the treatment of an acute migraine with or without aura (a sensory phenomenon or visual disturbance) in adults. However, it is not to be used prophylactically. In the US, it is marketed under the brand name, Nurtec ODT.[1]
It is not indicated for the preventive treatment of migraine.[1] It is taken by mouth, to dissolve on the tongue.[1] It takes effect within an hour and can provide relief for up to 48 hours, according to Biohaven. It is not a narcotic and has no addictive potential, and consequently will not be designated a controlled substance. It works by blocking CGRP receptors. 86% of patients did not require additional rescue medication within 24 hours of a single dose of Nurtec. All this info was obtained from a press release from Biohaven. (https://www.prnewswire.com/news-releases/biohavens-nurtec-odt-rimegepant-receives-fda-approval-for-the-acute-treatment-of-migraine-in-adults-301013021.html)
Rimegepant was approved for use in the United States as of February 27th, 2020 by the U.S. Food and Drug Administration (FDA) to be produced and marketed by Biohaven Pharmaceuticals.[2]
Mechanism of action
Rimegepant is a small molecule calcitonin gene-related peptide (CGRP) receptor antagonist.[3]
PATENTS
WO 2011046997
PATENT
WO 2012050764
https://patents.google.com/patent/WO2012050764A1
The disclosure generally relates to a synthetic process for preparing compounds of formula I including the preparation of chemical intermediates useful in this process. CGRP inhibitors are postulated to be useful in pathophysiologic conditions where excessive CGRP receptor activation has occurred. Some of these include neurogenic vasodilation, neurogenic inflammation, migraine, cluster headache and other headaches, thermal injury, circulatory shock, menopausal flushing, and asthma. CGRP antagonists have shown efficacy in human clinical trials. See Davis CD, Xu C. Curr Top Med Chem. 2008 8(16):1468-79; Benemei S, Nicoletti P, Capone JG, Geppetti P. Curr Opin Pharmacol 2009 9(1):9-14. Epub 2009 Jan 20; Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, Leibensperger H, Froman S, Assaid C, Lines C, Koppen H, Winner PK. Lancet. 2008 372:2115. Epub 2008 Nov 25; Ho TW, Mannix LK, Fan X, Assaid C, Furtek C, Jones CJ, Lines CR, Rapoport AM; Neurology 2008 70: 1304. Epub 2007 Oct 3.
CGRP receptor antagonists have been disclosed in PCT publications WO 2004/092166, WO 2004/092168, and WO 2007/120590. The compound (5S,6S,9R)- 5-amino-6-(2,3-difluorophenyl)-6,7,8!9-tetrahydiO-5H-cyclohepta[b]pyridin-9-yl 4- (2-oxo-2,3-dihydiO-lH-imidazo[4,5-b]pyridin-l-yl)piperidine-l-carboxylate is an inhibitor of the calcitonin gene-related peptide (CGRP) receptor.


cheme 1 illustrates a synthesis of formula I compounds. heme 1,

DESCRIPTION OF SPECIFIC EMBODIMENTS
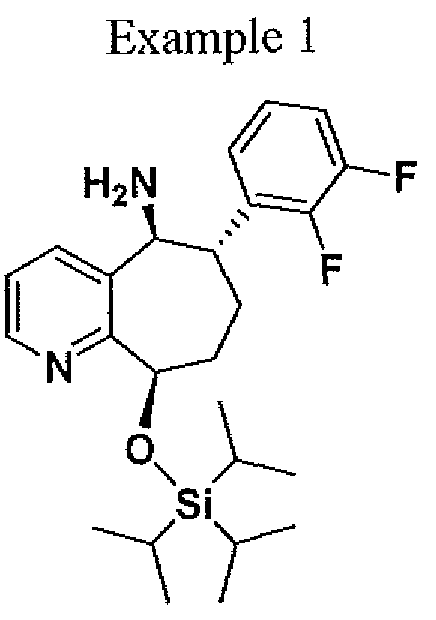
( 6S, 9R)-6~ (2, 3 -difluorophenyl)-9-(triisopropylsiIyloxy) – 6, 7, 8, 9-tetrahydro-5H- cyclohepta[b]pyridin-5 -amine. To a 100 mL hastelloy autoclave reactor was charged (6S,9R)-6-(2,3-difluorophenyl)-9-(triisopiOpylsilyloxy)-6,7,8,9-tetrahydi -5H- cyclohepta[b]pyridin-5-one (5.00 g, 1 1.22 mmol), 1,4-dioxane (50 mL) and titanium tetra(isopropoxide) (8.33 mL, 28.11 mmol). The reactor was purged three times with nitrogen and three times with ammonia. After the purge cycle was completed, the reactor was pressurized with ammonia to 100 psig. The reaction mixture was heated to 50°C (jacket temperature) and stirred at a speed to ensure good mixing. The reaction mixture was aged at 100 psig ammonia and 50°C for 20 h. The mixture was then cooled to 20°C then 5 % Pd/Alumina (1.0 g, 20 wt%) was charged to the autoclave reactor. The reactor was purged three times with nitrogen and three times with hydrogen. After the purged cycle completed, the reactor was pressurized with hydrogen to 100 psig and mixture was heated to 50°C (jacket temperature) and stirred at a speed to ensure good mixing. The reaction mixture was aged at 100 psig H2 and 50°C for 23h (reactor pressure jumped to -200 psig due to soluble ammonia in the mixture). The mixture was then cooled to 20 °C then filtered then transferred to a 100 ml 3-necked flask. To the mixture water (0.55 mL) was added drop wise, which resulted in yellow slurry. The resulting slurry was stirred for 30 mm then filtered, then the titanium dioxide cake was washed with 1,4-dioxane (30 mL). The filtrate was collected and the solvent was removed. The resulting oil was dissolved in isopropanol (40 mL). To the solution ~5N HC1 in isopropanol (9.0 ml) was added drop wise resulting in a thick slurry. To the slurry isopropyi acetate (60 ml) was added and heated to 45 °C for 10 min and then cooled to 22 °C over approximately 3 h to afford a white solid (3.0 g, 51.5 %). Ή NMR (500 MHz, CD3OD)
δ ppm 8.89 (d, J= 5.3, 1H), 8,42 (bs, 1H), 8.05 (bs, 1H), 7.35 (dd, J= 8.19 , 16.71), 7.2 (bs, 2H), 7.22 (m, 1H) 7.15 (m, 1H), 5.7 (dd, J = 1.89, J = 8.51), 5.4 (m, 1H), 3.5 ( m, 1H), 1.9-2.5 (B, 4h) 1.4 (sept, J = 15.13,3H), 1.2 (t, J= Ί.5Ί 18H); 13C NMR (125 MHz, CD3OD) δ 153.5, 151.6, 151.5, 151.3, 149.4, 143.4, 135.03, 129.8, 129.8, 127.8, 126.8, 126.4, 118.6, 72.4, 54.1, 41.4, 34.3, 32.3, 25.4, 18.6, 18.5, 13.7, 13.6, 13.5, 13.3.
Example 2

(6S,9R)-5-cmino-6-(2 -difluorophenyl)-6, 7,8,9-tetrahydro~5H-cyclohepta[b^ 9-o To a 250 ml flask was charged (6S,9R)-6-(253-difluoiOphenyl)-9-
(tnisopiOpylsilyloxy)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-amine di HC1 salt (15 g, 25.88 mtnol) and a solution of isopropanol: water (45 mL : 15 mL). The mixture was heated to 82 °C for 6h then dried via azeotropic distillation at atmospheric pressure using isopropanol until the KF was less than < 3 %. After fresh isopropanol (25 ml) was added, the mixture was heated to 70 °C and then isopropyl acetate (45 ml) was added that resulting in a white slurry. The slurry cooled to 22 °C for 15 min to afford a white solid (9.33 g, 99%). 1H NMR (500 MHz CD3OD) δ 8.77 (d, J= 5.7 Hz, 1H), 8.47 (d, J= 7.9 Hz, 1H), 8.11 (dd, J= 6.0, 8.2 Hz, 1H), 7.21-7.32 (m, 3H), 5.53 (dd, J= 3.8, 9.8 Hz, 1H) 5.33 (d, J = 9.8 Hz, 1H), 3.5 (bm, 1H), 2.25- 2.40 (m, 2H), 2.15 (bm, 1H), 1.90 (bm, 1H); 13C NMR (125 MHz, MeOD) δ 159.4, 153.9, 151.9 and 151.8, 149.7, 143.6, 141.8, 135.7, 130.6, 127.7, 126.8, 1 18.9, 70.0, 54.9, 42.2, 34.5, 33.4. Example 3

(5S, 6S, 9R)-5-amino-6-(2, 3-difluorophenyl)-6, 7>8,9-tetrahydro-5H- cyclohepta[b ]pyridin-9~yl~4-(2-oxo-2, 3-dihydro-lH-imidazo[4, 5-b ]pyridin-l- yl)piperidine-l-carboxylate. To a round bottom flask was charged (5S,6S,9R)-5- amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-ol dihydrochloride (1.00 g, 2.73 mmol) and dichloromethane (15 mL). A solution of sodium carbonate (0.58 g, 5.47 mmol), 20 wt% aqueous sodium chloride (5 mL), and water (10 mL) was added and the biphasic mixture was aged for 30 min. The phases were allowed to separate and the organic stream was retained. The dichloromethane solvent was then switched with azeotropic drying to tetrahydrofuran, with a final volume of (15 mL). At 20 °C was added, l-(l-(lH~imidazole-l-carbonyl)piperidin- 4-yl)-lH-imidazo[4,5-b]pyridin-2(3H)-one (0.95 g, 3.01 mmol), followed by a 20 wt% potassium ter/-butoxide solution in THF (4 mL, 6.20 mmol). The thin slurry was aged for lh, and then the reaction was quenched with the addition of 20 wt% aqueous sodium chloride (5 mL) and 20 wt% aqueous citric acid (2.5 mL). The layers were allowed to separate and the organic rich layer was retained. The organic layer was washed with 20 wt% aqueous sodium chloride (1 mL). The organic tetrahydrofuran stream was then concentrated in vacuo to afford an oil which was resuspended in dichloromethane (20 mL) and dried with MgS04. The
dichloromethane stream was concentrated in vacuo to afford an oil, which was crystallized from ethanohheptane to afford a white solid (1.14 g, 78.3%). LCMS: [M+H] = 535: 1H MR (600 MHz, 6-DMSO) δ 11.58 (IH, bs), 8.45 (IH, bd), 8.03 (IH, d, J= 7.3 Hz), 7.91 (IH, bs), 7.54 (IH, bd), 7.36 (IH, bm), 7.34 (IH, bm), 7.28 (IH, m), 7.21 (IH, m), 7.01 (IH, bs), 6.01 (IH, dd, J= 3.2, 9.8 Hz), 4.48 (IH, d, J= 9.5 Hz), 4.43 (IH, bm), 4.38 (IH, bm), 4.11 (IH, bm), 3.08 (IH, bm), 2.93 (IH, bm), 2.84 (IH, m), 2.62 (IH, bm), 2.20 (2H, bm), 2.13 (IH, bm), 2.12 (IH, bm), 1.75 (IH, bm), 1.72 (1H, bm), 1.66 (1H, bm); C NMR (125 MHz, i/6-DMSO) δ 156.6, 154.2, 153.0, 149.8, 148.1, 146.4, 143.5, 139.6, 137.4, 134.0, 132.8, 124.7, 124.5, 123.3, 122.2, 116.3, 115.0, 114.3, 73.7, 52.8, 50.0, 43.8, 43.3, 32.0, 30.3, 28.6; nip 255°C.
Example 4

l-(l-(lH^mdazole-l-carbonyl)piperidin-4-yl)-lH-imidazo
To a round bottom flask was added, Ι,Γ-carbonyldiimidazole (8.59 g, 51.4 mmoi), diisopropylethylamine (12.6 mL, 72.2 mmol) and tetrahydrofuran (100 niL). This mixture was warmed to 40°C and aged for 10 min, after which l-(piperidin-4-yl)-lH- imidazo[4,5-b]pyridin-2(3H)-one dihydrochloride (10 g, 34,3 mmol) was added. The slurry was aged at 40 °C for 3 h, and then upon reaction completion, the solvent was swapped to acetonitrile which afforded an off white solid (9.19 g, 85.9%). LCMS: [M+H] = 313; Ή NMR (400 MHz, 6-DMSO) δ 11.58 (1H, s), 8.09 (1H, s), 7.97 (1H, d, J= 8.0 Hz), 7.73 (1H, d, J= 4.0 Hz), 7.53 (1H, s), 7.05 (1H, s), 7.00 (1H, dd, J= 4.0, 8.0 Hz), 4.52, (1H, dd, J= 8.0, 12.0 Hz), 4.05 (2H, bd, J= 8,0 Hz), 3.31 (2H, m), 2.34 (2H, m), 1.82 (2H, bd, J = 12.0 Hz); 13C NMR (100 MHz, i/6~DMSO) δ 153.0, 150.4, 143.4, 139.8, 137.2, 128.9, 123.0, 1 18.7, 116.4, 115.2, 49.3, 45.1 , 28.5; mp 226°C.
Example 5

l-(l-(lH-imidazole-l-carbonyl)piperidin-4-yl)-lH-imidazo[4,5
To a 250 ml round bottom flask was added 3-N-piperidin-4-ylpyridine-2, 3 -diamine dihydrochloride (10 g, 52 mmol) and acetonitrile (100 mL). Triethyl amine (11.44 g, 1 13 mmol) and 1 , -Carbonyldiimidazole (18.34 g, 113 mmol) were added at ambient temperature and the mixture was stirred for 2 h. The solvent was evaporated under vacuum to—30 ml reaction volume and isopropyl acetate (50 mL) was added into the resulting sluny at 40°C. The slurry was cooled to 10-15 °C and then stirred for 1 h to afford an off white solid (10 g, 85%).
PATENT
US 20130225636
EP 2815749
PAPER
Journal of Medicinal Chemistry (2012), 55(23), 10644-10651.
https://pubs.acs.org/doi/full/10.1021/jm3013147

Calcitonin gene-related peptide (CGRP) receptor antagonists have demonstrated clinical efficacy in the treatment of acute migraine. Herein, we describe the design, synthesis, and preclinical characterization of a highly potent, oral CGRP receptor antagonist BMS-927711 (8). Compound 8 has good oral bioavailability in rat and cynomolgus monkey, attractive overall preclinical properties, and shows dose-dependent activity in a primate model of CGRP-induced facial blood flow. Compound 8 is presently in phase II clinical trials.
PAPER
Organic letters (2015), 17(24), 5982-5.
https://pubs.acs.org/doi/full/10.1021/acs.orglett.5b02921

An asymmetric synthesis of novel heterocyclic analogue of the CGRP receptor antagonist rimegepant (BMS-927711, 3) is reported. The cycloheptane ring was constructed by an intramolecular Heck reaction. The application of Hayashi–Miyaura and Ellman reactions furnished the aryl and the amine chiral centers, while the separable diastereomeric third chiral center alcohols led to both carbamate and urea analogues. This synthetic approach was applicable to both 6- and 5-membered heterocycles as exemplified by pyrazine and thiazole derivatives.

History
Originally discovered at Bristol-Myers Squibb,[4] it was under development by Biohaven Pharmaceuticals and is now also being marketed in the US by the same company after receiving FDA approval late February 2020.[5]
References
- ^ Jump up to:a b c “Nurtec ODT (rimegepant) orally disintegrating tablets, for sublingual or oral use” (PDF). February 2020. Retrieved 27 February 2020.
- ^ “Nurtec ODT: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 28 February 2020.
- ^ Diener HC, Charles A, Goadsby PJ, Holle D (October 2015). “New therapeutic approaches for the prevention and treatment of migraine”. The Lancet. Neurology. 14 (10): 1010–22. doi:10.1016/S1474-4422(15)00198-2. PMID 26376968.
- ^ “Rimegepant – Biohaven Pharmaceuticals Holding Company”. Adis Insight. Springer Nature Switzerland AG.
- ^ “Rimegepant (BHV-3000) – for acute treatment of Migraine”. Biohaven Pharmaceuticals.
External links
- “Rimegepant”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03461757 for “Trial in Adult Subjects With Acute Migraines” at ClinicalTrials.gov
|
|
| Clinical data | |
|---|---|
| Trade names | Nurtec ODT |
| Other names | BHV-3000, BMS-927711 |
| License data |
|
| Routes of administration |
By mouth |
| Drug class | calcitonin gene-related peptide receptor antagonist |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C28H28F2N6O3 |
| Molar mass | 534.568 g·mol−1 |
| 3D model (JSmol) | |
//////////Rimegepant , リメゲパント硫酸塩, Rimegepant sulfate, migraine, BMS-927711, fda 2020