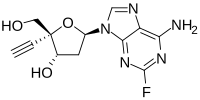
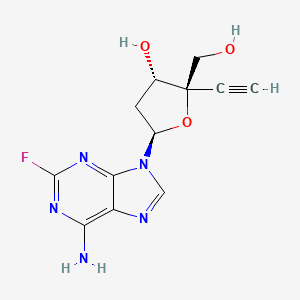
Islatravir (MK-8591, EFdA)
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
- Molecular FormulaC12H12FN5O3
- Average mass293.254 Da
- 865363-93-5
Islatravir is known to be a nucleoside reverse transcriptase inhibitor, useful for treating HIV-1 and -2 infection and AIDS.
Islatravir (MK-8591, EFdA), useful for the treatment of eg HIV, AIDS and related diseases.
Merck & Co and Idenix , under license from Yamasa Shoyu , are developing islatravir, a nucleoside reverse transcriptase inhibitor, for the oral prevention and treatment of HIV-1 and HIV-2 infection; in July 2019, data from a phase IIb trial in patients with HIV-1 infection were presented.In August 2015, Merck licensed Codexis ‘ CodeEvolver® protein engineering platform technology to develop enzymes for use in the manufacture of the pharmaceutical products such as islatravir.
Islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) is an investigational drug for the treatment of HIV infection.[1]It is classified as a nucleoside reverse transcriptase translocation inhibitor (NRTTI).[2] Merck is developing a subdermal drug-eluting implant to administer islatravir.[3][4]
Biological activity
Islatravir has activity against HIV in animal models,[5] and is being studied clinically for HIV treatment and prophylaxis.[6] Islatravir is a nucleoside analog reverse transcriptase translocation inhibitor that unlike other such inhibitors, inhibits HIV through multiple mechanisms,[5] providing rapid suppression of the virus, when tested in macaques and mice.[7] Nevertheless, there are HIV strains resistant to islatravir and research is ongoing.[8]
PATENTS
WO2020014046 ,
PATENT
WO2020014047
PATENT
WO2020014050 (assigned to Codexis ), covering engineered phosphopentomutase (PPM) enzymes, useful in the synthesis of pharmaceutical compounds including islatravir.
PATENT
WO-2020014041
4’-Ethynyl-2’-deoxy nucleoside analogs are known for activity against HIV, AIDS and related diseases.
One example of a 4’-ethynyl nucleoside analog is 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA, also known as MK-8591) which is a nucleoside reverse transcriptase translocation inhibitor that blocks HIV-l and SIV viral replication in vitro (Kawamoto, A., Kodama, E., Sarafianos S. F. et al, Int. J. Biochem. Cell Biol.; 40(l l):24lO-2O [2008]; Ohrui, H., Kohgo, S., Hayakawa, H. et al, Nucleosides, Nucleotides & Nucleic Acids, 26, 1543-1546
[2007]) and in vivo (Hattori, S., Ide, K., Nakata, H. et al. Antimicrobial. Agents and
Chemotherapy, 53, 3887-3893 [2009]). EFdA is claimed in US Patent No. 7,339,053 (referred to in the‘053 patent as 2,-deoxy-4’-C-ethynyl-2-fluoroadenosine). EFdA has the following chemical structure:
EFdA is metabolized in cells to its active triphosphate anabolite which inhibits HIV reverse transcriptase. In contrast to nucleoside reverse transcriptase inhibitors (NsRTIs) and nucleotide reverse transcriptase inhibitors (NtRTIs) currently available for the treatment of HIV infection which lack a 3′-OH group to block incorporation of incoming nucleotide, EFdA retains a 3′ OH group and acts as a chain terminator by preventing translocation of the primer template in the reverse transcriptase (RT) active site and preventing binding of incoming
deoxyribonucleotide triphosphates (dNTPs). In addition, the pucker of the modified ribose ring of EFdA is believed to contribute to inhibition of reverse transcriptase by placing the 3′-OH in a vector in which phosphotransfer from the incoming nucleotide is inefficient. (Michailidis E, et ak, Mechanism of inhibition of HIV-l reverse transcriptase by 4’-ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, J Biol Chem 284:35681-35691 [2009]; Michailidis E, et ak, 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-l reverse transcriptase with multiple mechanisms, J Biol Chem 289:24533-24548 [2014] ).
In in-vitro HIV replication assays, EFdA is a potent antiretroviral and exhibits comparable antiviral activity against clinical isolates across all subtypes that have been evaluated. It is rapidly anabolized in both lymphoid derived cell lines and in peripheral blood mononuclear cells to the active triphosphate in vitro, and the intracellular half-life of EFdA Triphosphate (EFdA- TP) exceeds 72 hrs. (Stoddart, C. A., Galkina, et ak, Oral Administration of the Nucleoside EFdA (4’-Ethynyl-2-Fluoro-2’-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque, Antimicrob Agents Chemother, 2015 Jul; 59(7): 4190-4198, Published online 2015 May 4).
EFdA has been shown to have efficacy in animal models of HIV infection including humanized mouse models and an SIV infected rhesus macaque model. Pharmacokinetic studies of orally administered EFdA in mouse and rhesus monkey have demonstrated rapid absorption and high plasma concentrations. A long intracellular half-life was demonstrated by the fact that isolated peripheral blood mononuclear cells from the rhesus macaque were refractory to SIV infection 24 hr after drug administration. (Ibid.)
Previous syntheses of 4’-ethynyl nucleoside analogs including EFdA suffer from modest stereoselectivity in the formation of the C-N bond between the ethynyl-deoxyribose sugar and the 2-fluoroadenine (also referred to as 2-fluoro-9H-purin-6-amine) nucleobase. The previous syntheses also require protecting groups to carry out the glycosylation reaction which reduces the efficiency of the syntheses.
The synthesis described in Kei Fukuyama, et ak, Synthesis of EFdA via a
Diastereoselective Aldol Reaction of a Protected 3-Keto Furanose, Organic Letters 2015, 17(4), pp. 828-831; DOI: 10.102 l/ol5036535) is a l4-step synthesis from D-glucose diacetonide that uses diastereoselective reactions to set the three stereocenters. The stereochemistry of the anomeric center is controlled by having a 2′-acetoxy directing group that is subsequently removed by hydrolysis and deoxygenation. This route requires 4 chromatographic purifications, and the stoichiometric use of a toxic organotin reagent for late-stage deoxygenation.
In another route (see Mark McLaughlin, et al., Enantioselective Synthesis of 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via Enzymatic Desymmetrization, Organic Letters 2017, 19 (4), pp. 926-929), the fully-substituted 4′- carbinol is generated stereoselectively with an enzymatic desymmetrization. The 3 ‘-stereocenter is set with a catalytic asymmetric transfer hydrogenation, and the anomeric 1 ‘-linkage is established in modest stereoselectivity using substrate control, with an upgrade in stereochemical purity achieved by crystallization of an intermediate. This process requires 15 steps, requires the use of several protecting groups and generates the glycosyl linkage between the nucleobase and sugar fragments in low
stereoselectivity (1.8: 1).
A l2-step synthesis for making EFdA from R-glyceraldehyde acetonide is described in Kageyama, M., et al., Concise Synthesis of the Anti-HIV Nucleoside EFdA, Biosci. Biotechnol. Biochem, 2012 , 76, pp. 1219 -1225; and Enantioselective Total Synthesis of the Potent Anti-HIV Nucleoside EFdA, Masayuki Kageyama, et al., Organic Letters 2011 13 (19), pp. 5264-5266 [DOL 10.1021 / ol202116k] . The syntheses use the chiral starting material to set the 3′-stereocenter with moderate diastereoselectivity. After chromatographic separation of stereoisomers, the new stereocenter is used to guide a diastereoselective alkyne addition to set the fully-substituted 4’-stereocenter. The anomeric 1 ‘-position is established with little stereocontrol and requires chromatography to separate the anomers. This route requires chromatographic separation of diastereoisomers at two different stages and starts from an expensive chiral starting material.
Kohgo, S., et al., Design, Efficient Synthesis, and Anti-HIV Activity of 4′-C-Cyano- and 4′-C-Ethynyl-2′-deoxy Purine Nucleosides, Nucleosides, Nucleotides and Nucleic Acids, 2004, 23, pp. 671-690 [ DOL 10.1081/NCN-120037508] describes a synthetic route that starts from an existing nucleoside and modifies both the sugar and nucleobase portions. It is an 18-step synthesis starting from 2-amino-2’-deoxy adenosine with a low 2.5% overall yield.
It is known that enzymes such as purine nucleoside phosphorylase (PNP, EC 2.4.2.1) can form the glycosyl linkage in nucleosides and nucleoside analogs in high stereoselectivity and without the use of protecting groups. See for example the review: New Trends in Nucleoside Biotechnology, Mikhailopulo, I. A., Miroshnikov, A.I,. Acta Naturae 2010, 2, pp. 36-58.
However, the current scope of the sugar fragments capable of undergoing reaction catalyzed by PNP has been limited to the a- 1 -phosphates of natural ribose and deoxyribose along with a small number of analogs with small H, NH2, or F substituents at the C2’ and C3’ positions and replacements of the C5’ OH group. There have been no reports of successful glycosylation catalyzed by PNP using sugars with carbon substituents on the ring or any substitution at the C4’ position.
Access to the ribose and deoxyribose a- 1 -phosphate substrates for the PNP-catalyzed glycosylation has been demonstrated by translocation of the phosphate group from the 5’-hydroxyl to G -hydroxyl position with the enzyme phosphopentomutase (PPM, EC 5.4.2.7) (see Mikhailopulo, I. A., et al. supra). However, the scope of the sugars for which PPM is capable of catalyzing this reaction has been limited to ribose, arabinose, 2-deoxyribose, and 2,3-dideoxyribose. No examples have been reported of successful reaction with sugar phosphates containing any additional substituents.
Deoxyribose phosphate aldolase (DERA, EC 4.1.2.4) enzymes are known to catalyze the aldol addition of acetaldehyde to other short-chain aldehydes (see review: Stephen M. Dean, et al., Recent Advances in Aldolase-Catalyzed Asymmetric Synthesis, Adv. Synth. Catal. 2007, 349, pp. 1308 – 1320; DOI: 10. l002/adsc.200700115). However, no examples have been reported with aldehydes bearing a fully substituted carbon a to the aldehyde.
ETS Patent 7,229, 797 describes the formation of deoxyribonucleosides from the natural unsubstituted deoxyribose 1 -phosphate by use of purine nucleoside phosphorylase (PNP) and additionally using enzymes such as sucrose phosphorylase to remove the inorganic phosphate byproduct and drive the equilibrium. It does not disclose enzyme engineering for the creation of PNP enzymes that can generate nucleosides from the unnatural 4-ethynyl-D-2-deoxyribose 1-phosphate, nor that through engineering of PPM and DERA enzymes to act on unnatural substrates, 4-ethynyl-D-2-deoxyribose 1 -phosphate can be generated.
In view of the difficult and lengthy synthetic options developed to date for producing 4’-ethynyl nucleoside analogs, it would be desirable to develop an improved enzymatic synthesis for 4’-ethynyl nucleoside analogs such as EFdA that reduces the number of process steps, minimizes the use of protecting groups, improves the stereoselectivity of glycosylation and avoids the use of toxic materials.
Surprisingly, it has been found that PPM enzymes have some activity with the 3-atom ethynyl substituent at the 4’ position on ribose and that the PPM enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a reaction for
isomerization of
4-ethynyl-D-2-deoxyribose 5-phosphate (6) to 4-ethynyl-D-2-deoxyribose 1 -phosphate (6.5) catalyzed by PPM to enable a more efficient method for production of 4’-ethynyl-2’-deoxy nucleosides.
Additionally, PNP enzymes have also been found to have some activity with the 3-atom ethynyl substituent at the 4 position on deoxyribose and that the PNP enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a glycosylation reaction catalyzed by PNP to enable a more efficient method for production of 4’ -ethynyl -2’-deoxy nucleosides.
Even further improvement to the overall synthetic method came from the finding that
DERA enzymes, particularly the DERA from Shewanella halifaxensis, have activity for aldol reaction with 2-ethynyl-glyceraldehyde 3-phosphate which has a fully substituted a-carbon. This discovery allowed for the efficient synthesis of 4-ethynyl-D-2-deoxyribose 5-phosphate, a precursor to 4’-ethynyl-2’-deoxy nucleoside analogs, e.g., including EFdA.
SUMMARY OF THE INVENTION
The present invention involves the use of engineered enzymes in a novel enzymatic synthesis of 4’-ethynyl-2’-deoxy nucleoside analogs, including EFdA, that eliminates the use of protecting groups on intermediates, improves the stereoselectivity of glycosylation and greatly reduces the number of process steps needed to make said compounds compared to prior methods, among other process improvements. It further relates to novel intermediates which are an integral part of the enzymatic process.
The overall process is summarized in the following Scheme 1 and Scheme 2; the latter scheme provides an alternative method for making compound 5:
Scheme 1
kinase
p p y
Scheme 1A
kinase galactose oxidase
3 2X+ 9
2
p p y
It has been discovered that 4’-ethynyl-2’-deoxy nucleoside analogs such as EFdA can be synthesized employing a final step one-pot process by combining 4-ethynyl-D-2-deoxyribose 5-phosphate (6) with two enzymes, phosphopentomutase (PPM) [for example but not limited to SEQ ID NO.: 8] and purine nucleoside phosphorylase (PNP) [for example but not limited to SEQ ID NO.: 9, SEQ ID NO.: 15], as shown in Scheme 2.
Scheme 2
Scheme 2A
Several upstream intermediates used in the present process for the synthesis of the final product 4’-ethynyl-2’-deoxy nucleosides and analogs thereof are also made using enzymatic reaction methods as shown in Scheme 3; Scheme 3 A and Scheme 3B
Scheme 3
Scheme 3A
o2
pTsOH
deoxyribose
aldolase
Scheme 3B
Experimental Procedures
Preparation of 2-ethynyl-2-hvdroxypropane-l,3-diyl diacetate 12)
Method A:
To a -35 °C solution of diacetoxyacetone (1) (159 g, 914.0 mmol) in THF (1000 mL) was added 1600 mL of a 0.5 M solution of ethynyl magnesium chloride in THF maintaining the temperature below -20 °C. After the reaction reached completion, acetic acid (78 mL) in 400 mL methyl tert-butyl ether (MTBE) was added dropwise keeping the temperature below -20 °C. MTBE (800 mL) was then added and the mixture was warmed to room temp. Saturated NaCl in water (1000 mL) was added followed by saturated NH4CI solution in water (1050 mL). The organic layer was separated, dried over Na2SC>4 and evaporated to give compound (2) as an oil (160 g, 88%). 1H NMR (CDCI3, 500 MHz): d 4.26 (dd, 4 H), 2.55 (s, 1H), 2.14 (s, 6H).
Preparation of 2-ethynyl-propane-l,2,3-triol 13)
Method B:
To a solution of 2-ethynyl-2-hydroxypropane-l,3-diyl diacetate (2) (70 g, 350 mmol) in ethanol was added a 0.5M solution of sodium methoxylate in methanol (69.9 mL, 35.0 mmol) at room temperature (rt). The reaction was stirred at rt for 2 hours (h) to reach completion. The solvents were evaporated and the residue was re-dissolved in 100 mL water and extracted with 3 x 50 mL MTBE. The aqueous layer was sparged with nitrogen to remove residual solvents to give a 40.9% solution of 2-ethynyl-propane-l,2,3-triol (3) (108 g , 100% yield) as determined by nuclear magnetic resonance (NMR) (maleic acid as internal standard). lH NMR (D2O, 500 MHz): d 3.60 (dd, 4 H), 2.85 (s, 1H).
Alternate Preparations o ethynyl-glvcer aldehyde 14)
Method Cl:
In a stirred reactor, 2-ethynyl-propane-l,2,3-triol (3) (1.1 g, 9.47 mmol) in sodium phosphate buffer (30 mL, 100 mM, pH 7.0) containing antifoam 204 (Sigma A6426, 1 drop ~ 20 pL) was warmed to 30 °C with air sparging at 12.5 seem. Galactose oxidase (GOase, SEQ ID NO.: 1) (250 mg), Horseradish Peroxidase* (Type I, 5 mg) and bovine catalase** (5 mg) dissolved in sodium phosphate buffer (5 mL 100 mM, pH 7.0) were added to the reactor, followed by the addition of CuS04 aq. solution (100 mM, 150 pL). The reaction mixture was stirred at 600 rpm with air sparging for 47h to give (f?)-2-ethynyl-glyceraldehyde (4) in 47% conversion (by NMR) and 72% e.e. . (The product was not isolated). lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish Type I, commercially available from SIGMA (P8125), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345)
Method C2:
In a stirred 100 L jacketed reactor charged with deionized water (56.2 kg), sodium dihydrogen phosphate (1.212 kg, 10 moles) was added. The pH was adjusted to 7.02 using 10 N sodium hydroxide solution (852.6 g) at 25 °C. The reactor was charged with Antifoam 204 (A6426, 10 mL), followed CuS04*5H20 (6.5 g). Galactose oxidase (451.2 g) (SEQ ID NO.: 10) was added and stirred for 15 min while sparged with air. Horseradish peroxidase* (200.2 g) and catalase** (502.6 g) were added and the reactor was rinsed with water (2.0 kg). Next 2-ethynyl-propane- 1,2, 3 -triol (3) solution in water (9.48%, 30.34 kg, 24.72 mol) was added followed by an additional portion of Antifoam 204 (A6426, 10 mL). The reaction was sparged with air and
stirred overnight to give 94.0 kg of (A)-2-ethynyl-glyceraldehyde (4) in 66% conversion (by NMR) and 84% e.e. Assay yield 60%: 1H NMR (D20, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish purified, commercially available from Toyobo (PEO-301), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345).
The above reaction was also performed using the galactose oxidase (SEQ ID NO.: 11) and the product (4) was obtained in 67% conversion (by NMR) and 88% e.e. and assay yield 59%: 1H NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
Method C3:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 0.5 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase (SEQ ID NO.: 17, 450 mg enzyme powder) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (Cl 345, Sigma-Aldrich, 150 mg, 2000-5000 U/mg, 0.75 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg,
130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (25 wt%, 12 mL, 25.8 mmol). The reaction mixture was stirred at 30 °C with aeration at 125 seem and sampled using EasySampler over 20h to give 70% conversion and form compound (4) ((A)- 2-ethynyl-glyceraldehyde) in 58% assay yield and 99% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C4: Oxidation with immobilized galactose oxidase
Galactose
Oxidase
immobilized
3
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (16 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 160 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution. In a vessel evolved galactose oxidase (SEQ ID NO.: 17, 2.00 g) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (50 mL) and the resin. The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 160 mL) and potassium PIPES buffer (10 column volumes, 160 mL; 50 mM, pH 7.5) and it was used directly in a reaction. Reaction procedure:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 1 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase immobilized on the resin (SEQ ID NO.: 17, 750 mg enzyme powder per 6 mL resin) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (C1345, Sigma-Aldrich, 210 mg, 2000-5000 U/mg, 1.05 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg, 130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane- 1,2, 3 -triol (3) (25 wt%, 13 mL, 29.4 mmol). The reaction mixture was stirred at 25 °C with aeration at 125 seem. After 22h the reaction reached 91% conversion to give 200 mM (//)-2-ethynyl-glyceraldehyde (4) solution (100 mL, 68% assay yield, 97% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C5: Optional Isolation of aldehyde via formation of aminal (8)
Step 1: Preparation of (S)-2-( \ .3-dibenzylimidazolidin-2-yl )but-3-yne- l 2-diol
A 100 L jacketed cylindrical vessel equipped with nitrogen bubbler, mechanical stirrer and thermocouple was charged with crude oxidase reaction stream containing (f?)-2-ethynyl-glyceraldehyde ((4), 26.0 kg, 1.85 wt% aldehyde, 3.64 mol) and inerted with N2 atmosphere. The aqueous solution was warmed to 20 °C and Af,A-di methyl dodecan- 1 -ami ne oxide (DDAO) (30 wt% in water, 798 g, 0.96 mol;) was added, followed by MTBE (55.3 kg, 76 L) and N,N -dibenzylethane-l, 2-diamine (1.55 kg, 6.43 mol). The brown, biphasic mixture was stirred overnight at 20 °C under nitrogen atmosphere. After 17 hours the stirring was stopped and the organic phase was removed and discarded. A light brown MTBE solution of fV)-2-( l ,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (56.5 kg, 2.02 wt% aminal, 3.39 mmol, 93% assay yield) was obtained.
Six similar MTBE solutions were processed together in a single distillation and crystallization step (in total 374.4 kg of solution, containing 7.91 kg aminal).
A 50 L jacketed cylindrical vessel equipped with mechanical stirrer, distillation head (condenser at -20 °C) and thermocouple was charged with aminal solution (45 L). Vacuum was applied to the vessel (65-95 torr) and the jacket was set to 40 °C. Solvent was removed by distillation until a volume of 35 L had been reached. At this point, the internal temperature was 6.1 °C and an off-white solid had begun to crystallize. The remaining MTBE solution was slowly added, maintaining a constant volume of 35-40 L and an internal temperature of 0-10 °C. Once all the MTBE solution had been added the volume was decreased to 25 L. Distillation was halted, the vessel was inerted with nitrogen and the jacket temperature was decreased to 10 °C. The resulting pale yellow suspension was aged at this temperature for 2 hours and the solids were collected by filtration. The filter cake was washed with cold (-2 °C) MTBE (12.7 kg) and then dried under nitrogen flow for 7 hours. (5)-2-(l,3-dibenzylimidazolidin-2-yl)-but-3-yne-l,2-diol was obtained as an off-white crystalline solid (5.75 kg) lff NMR (500 MHz, DMSO-i¾) d 7.42 – 7.35 (m, 4H), 7.32 (td, J= 7.5, 1.6 Hz, 4H), 7.27 – 7.21 (m, 2H), 5.10 (t, J= 5.6 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J= l3.3Hz, 1H), 4.16 (d, J= 13.3 Hz, 1H), 3.76 (s, 1H), 3.70 – 3.58 (m, 4H), 3.21 (d, J= 0.9 Hz, 1H), 2.90 – 2.80 (m, 2H), 2.60 – 2.51 (m, 2H).13C NMR (126 MHz, DMSO-i¾) d 140.0, 140.0, 128.5, 128.3, 128.2, 128.1, 126.8, 126.8, 88.6, 86.9, 75.0, 74.0, 66.4, 60.7, 60.5, 50.4, 50.3, 39.5. HR-MS (ESI) Aminal (M + H+) C21H25N202+ calculated 337.1911; found 337.1922.
Step 2: Prep l (8)
A 4 L jacketed cylindrical vessel equipped with nitrogen bubbler and mechanical stirrer was charged with of TsOH»H20 (12.0 g, 63.1 mmol), water (60 mL), (ri)-2-(l,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (110 g, 327 mmol) and MTBE (1700 mL). The biphasic mixture was placed under nitrogen and the jacket temperature was set to 15 °C. A solution of TsOH»H20 (114 g, 599.3 mmol) in water (600 mL) was added dropwise over 1.5 hours with overhead stirring (200 rpm). After addition had completed, the jacket temperature was lowered to 5 °C and the resulting slurry was aged for 1 hour. The solids were removed by filtration and washed with cold water (270 mL). The biphasic solution was transferred to a separating funnel and the organic phase was removed and discarded. The aqueous phase was treated with DOWEX MARATHON A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
MARATHON A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
(ESI) Aldehyde dimer (2M + Na+) CioHi2Na06+ calculated 251.0526; found 251.0530.
Alternate Preparations o ethvnyl-glvceraldehvde 3-phosphate (5):
Method Dl: Acetate kinase: ATP -regeneration system
Pantothenate kinase PanK
ATP
Acetate kinase
4 Acetate phosphate
5
In a stirred reactor, to a solution of adenosine diphosphate disodium salt (40 mg, 0.087 mmol) and magnesium chloride (38 mg, 0.400 mmol) in HEPES buffer (66 mM, pH 7.5, 30 mL) was added (i?)-2-ethynyl-glyceraldehyde (4) (1.9 mL, 210 g/L solution in water, 3.51 mmol), followed by acetate kinase (SEQ ID NO.: 3) (40 mg), and pantothenate kinase (SEQ ID NO.: 2) (120 mg). The reaction mixture was warmed to 25 °C and a solution of acetyl phosphate lithium potassium salt (1.3 g, 7.01 mmol) in HEPES buffer (50 mM, pH 7.5, 10 mL) was added dropwise over 4 hours, with pH maintained at 7.5 using 5M sodium hydroxide. The reaction was stirred for 18 hours to give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 85% conversion (by HPLC) (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method D2: Pyruvate oxidase ATP -regeneration system
Pan
Pyruvate oxidase
Pyruvate
Phosphate
02
In a stirred reactor, a solution of sodium pyruvate (3.11 g, 28 mmol) and phosphoric acid (0.523 mL, 7.71 mmol) in 76 mL water pH 7.5 was charged with (i?)-2-ethynyl-glyceraldehyde (4) (3.8 mL, 210 g/L solution in water, 7.01 mmol), adenosine diphosphate disodium salt (80 mg, 0.174 mmol), thiamine pyrophosphate (40 mg, 0.086 mmol), flavin adenine dinucleotide disodium salt hydrate (64 mg, 0.077 mmol), and magnesium chloride (400 pL, 1 M solution in water, 0.4 mmol). The pH was re-adjusted to 7.5 with 5M aq sodium hydroxide and the reaction volume was re-adjusted to 80 mL with water. Acetate kinase (SEQ ID NO.: 3) (80 mg), pyruvate oxidase (SEQ ID NO.: 4) (80 mg, lyophilized cell free extract), pantothenate kinase (SEQ ID NO.: 2) (400 mg), and catalase (800 pL, ammonium sulfate suspension CAT-101, Biocatalytics) were added. The reaction was stirred at 500 rpm and 30 °C with air sparging for 72 hours to give (//)-2-ethynyl-glyceraldehyde 3 -phosphate 5 in 95% conversion (by HPLC) (The product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
The above reaction was also performed using the pantothenate kinase (SEQ ID NO.: 13) and the product 5 was obtained in 66% conversion. (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H).
Method D3: Acetate kinase: ATP -regeneration system using immobilized enzymes
Panth
Acetate phosphate
Enzyme immobilization procedure:
NUVIA Immobilized Metal-ion Affinity Chromatography (IMAC) nickel-charged resin (168 mL based on settled volume) was added to a filter funnel and washed with binding buffer (1.6 L; 500 mM sodium chloride, 50 mM sodium phosphate, pH 8.0). In a vessel, pantothenate kinase
(8.4 g) (SEQ ID NO.: 12) and acetate kinase (2.8 g) (SEQ ID NO.: 3) were dissolved in binding buffer (500 mL). The washed resin was charged to the vessel and the solution was stirred for 4 hours at 20 °C. The resin was filtered and washed first with binding buffer (1.6 L) followed by piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (840 mL; 50 mM, pH 6.5). The washed resin was used directly in the next step.
Reaction procedure:
To a 1 L reactor, a solution of (f?)-2-ethynyl-glyceraldehyde (4) in water (608.7 g, 4.6 wt%, 212 mmol) was charged and cooled to 5 °C. To the cooled solution piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (32.7 mL, 1 M, pH 6.5, 32.7 mmol), magnesium chloride (9.33 mL, 1 M, 9.33 mmol), acetyl phosphate diammonium salt (51.8 g, 265 mmol), adenosine diphosphate disodium salt hydrate (1.17 g, 2.12 mmol), and water (192 mL) were added. The solution was allowed to stir and pH was adjusted to 6.4 using 5 N KOH. The reaction was warmed to 20 °C and 168 mL of resin with co-immobilized pantothenate kinase (SEQ ID NO.: 12) and acetate kinase (SEQ ID NO.: 3) was added. The reaction was stirred for 10 hours with 5 N KOH used to maintain a pH of 6.4 to give (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in
92% conversion (by HPLC) and 91% yield (by 3 lp NMR with tetraphenylphosphonium chloride as internal standard) (the product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Preparation of 4-ethynyl-D-2-deoxyribose 5-phosphate 16)
Method E:
To a solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (5, 20 mL, 5.3 mmol) in water, a solution of acetaldehyde in water (40 wt.%, 2.02 mL, 15.9 mmol) was added at room
temperature, followed by the addition of Deoxyribose-phosphate aldolase (DERA) (SEQ ID NO. : 6), 25 mg solution in triethanolamine hydrochloride buffer (1 mL, 1 M, pH 7.0). The reactor was sealed and the mixture was stirred overnight at 30 °C and 600 rpm to give 4-ethynyl-D-2-deoxyribose 5-phosphate (6) in 99% conv. and 99% e.e., 99% d.e. as a 1 : 1 anomer mixture (The product was not isolated) a-anomer: lH NMR (D2O, 600 MHz) 5 5.31 (t, 1H), 4.13 (t, 1H), 3.81-3.72 (m, 2H), 2.89 (s, 1H), 2.42-2.34 (m, 1H), 1.87-1.79 (m, 1H); 13c NMR (D2O, 151 MHz) 5 97.7 (s), 81.4 (d), 79.4 (s), 78.9 (s), 71.1 (s), 67.7 (d), 39.6 (s). b-anomer: 1H NMR
(D2O, 600 MHz) 5 5.40 (dd, 1H), 4.28 (t, 1H), 3.88-3.80 (m, 2H), 2.87 (s, 1H), 2.13-2.06 (m,
1H), 2.04-1.97 (m, 1H); 13C NMR (D20, 151 MHz) 5 97.3 (s), 82.2 (d), 78.7 (s), 78.5 (s), 71.3 (s), 68.4 (d), 39.6 (s). LC-MS: (ES, m/z): calculated for C7H10O7P (M-H): 237.0; found 237.0
Alternate Preparations of (2 ?,3A,5 ?)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol monohydrate (7) [alternative name 4’-ethynyl-2-fluoro- 2’-deoxvadenosine or EFdAI
Method FI:
Ammonium ((2f?,3ri)-2-ethynyl-3,5-dihydroxytetrahydrofuran-2-yl)m ethyl hydrogen phosphate (1.00 g, 3.91 mmol) was dissolved in 10 mL of pH 7.5 buffer (100 mM triethanolamine ΉO containing 5 mM MnCl2). The solution pH was adjusted to 7.3 with 5 N NaOH. To the solution was added 2-fluoroadenine (0.599 g, 3.91 mmol) and sucrose (2.68 g, 7.82 mmol). The enzyme solution was prepared by dissolving phosphopentomutase (SEQ ID NO. : 8) (100 mg), purine nucleoside phosphorylase (SEQ ID NO.: 9) (50 mg), and sucrose phosphorylase (SEQ ID NO. :
7) (10 mg) in 10 mL of the pH 7.5 buffer. The enzyme solution was added to the reagent mixture and the resulting suspension was shaken at 40 °C. After 20 h, the suspension was cooled to 0 °C and filtered, rinsing with cold water. The solid was suction dried to give the title compound (1.12 g, 92%) as a single isomer.
iH NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J =
5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13c NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
The PPM and PNP enzymes used in this step were each derived from mutations starting from the enzymes from E. coli ( Escherichia coli). The sucrose phosphorylase (SP) used in this step was derived from Alloscardovia omnicolens ; SP derived from other organisms could also be used.
Method F2:
To an aqueous solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (950 mL, 157 mmol) containing piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at a pH from about 5.5 to 6.0 was added triethanolamine (7.09 g, 47.5 mmol). The pH of the solution was adjusted from 7.1 to 7.6 using potassium hydroxide (8 mL, 8M). Manganese(II) chloride hydrate (0.592 g, 4.70 mmol) was added followed by sucrose (161 g, 470 mmol), giving a pH of 7.5 To the solution
was added the following enzymes: deoxyribose-phosphate aldolase (SEQ ID NO. : 14) (461 mg), sucrose phosphorylase (SEQ ID NO. : 7) (494 mg), phosphopentomutase (SEQ ID NO.: 8)(2.63 g), and purine nucleoside phosphorylase (SEQ ID NO. : 15) (659 mg). Once the enzymes were dissolved, 2-fluoroadenine (19.80 g, 125 mmol) was added. The reaction was heated to 35 °C and acetaldehyde was added (40 wt% in isopropyl alcohol, 29.8 mL, 235 mmol). After reacting for 2h, the mixture was seeded with EFdA crystalline product (0.96 g, 2 mol%). After reacting over 26 h at 35 °C, the slurry was cooled to 0 °C, and the solids were collected by filtration, washing with water two times (40 mL ea.). The solids were dried under a nitrogen sweep. Yield 43.2 g, 92 wt%, 96.2% corrected. ¾ NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13C NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
Alternate Preparations of -2-ethvnyl-propane-l,2,3-triol 1 1-phosphate 19) :
Method Gl: Acetate kinase: ATP-regeneration system using enzymes SEQ. ID No.: 2 and SEQ. ID No.: 3
Panthotenate kinase PanK
ATP
Acetate kinase
Acetate phosphate
A 50 mL reactor was charged with a solution of 2-ethynyl-propane-l,2,3-triol (3) in water (9.29 g, 9.46 wt%, 7.57 mmol) potassium PIPES buffer (1.02 mL, 1 M, pH 6.5, 1.02 mmol), magnesium chloride (292 pL, 1 M, 0.292 mmol), acetyl phosphate diammonium salt (1.851 g, 89 wt%, 9.46 mmol), adenosine diphosphate disodium salt hydrate (ADP, 42 mg, 0.076 mmol, 0.01 eq), and water (28 mL). The pH was adjusted to 6.4 using 5 M KOH, the solution was warmed to 20 °C and evolved pantothenate kinase PanK SEQ. ID No.: 2 (264 mg) and acetate kinase AcK SEQ. ID No. : 3 (88 mg) were added. The reaction was stirred for 16 hours with pH maintained at 6.4 using 5 N KOH. The final reaction contents provided C.V)-2-ethynyl -propane- 1 ,2,3-triol 1-phosphate (9) in >95% e.e. and 99% conversion (by 31P NMR). The product was not isolated. ¾ NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H). 13C NMR (D2O, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s). 31P NMR (D2O, 202 MHz) d 3.39. HRMS: (ESI, m/z): calculated for [M-l] CsHsOeP: 195.0058; Found 195.0068 [M-H] : 195.0058.
Method G2: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
To a jacketed reactor aqueous solution 2-ethynyl-propane-l,2,3-triol (3) (11.47 kg, 8.7% wt, 8.61 mol) and water (7.5kg) was charged, followed by 1M BIS-TRIS methane buffer pH 6.5 (1L) and magnesium chloride (41.4 g). ATP (48g, 0.086 mol, 0.01 equivalent) and diammonium acetyl phosphate (2.021 kg, 89%, 10.33 mmol) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using KOH (270.4 g). Evolved pantothenate kinase SEQ. ID No.: 20 (20.4 g) and evolved acetate kinase SEQ. ID No.: 21 (3 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which pH dropped to 5.5.
Quantitative conversion of 2-ethynyl-propane-l,2,3-triol (3) was obtained as judged by ‘H and 31P NMR. Such prepared (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) solution (397 mM, 22.5 kg, 98% yield) was used in subsequent oxidation step without any further purification. ‘H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H).
Method G3: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21 with deuterated compound (3) to assign absolute stereochemistry and demonstrate desymmetrizing phosphorylation.
Acetate phosphate
Z-d2, 95:5 er
Evolved pantothenate kinase SEQ. ID No. : 20 (100 pL of 10 g/L solution in water ) and evolved acetate kinase SEQ. ID No. : 21 (100 pL of 2g/L solution in water) were added to a solution containing diammonium acetyl phosphate (41 mg), 2-ethynyl-propane-l, l-72-l,2,3-triol ((A)- 3-d2, 20 mg, 170 pmol), magnesium chloride (10 pL of 1 M solution in water), ADP (10 pL of 100 g/L solution in water), and sodium phosphate buffer (10 pL of 1 M solution in water) in water (800 pL) at pH 6.5. The reaction was incubated for 24h at rt to give deuterated 2-ethynyl-propane-l,2,3-triol l-phosphate analogs (S)-9-(3,3-d2) and (S)-9-(l,l-d2) in 95:5 ratio and 99% overall yield. The ratio of phosphorylated compounds was determined by 31P NMR to be -95:5, confirming stereoselective phosphorylation of the 2-ethynyl-propane-l,2,3-triol (3) at the pro-(S) hydroxyl group (i.e. a desymmetrizing phosphorylation). 1H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H), 2.93 (s, 1H). 13C NMR (D20, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s).
Method G4: Acetate kinase: ATP-regeneration system using immobilized enzymes SEQ. ID No. : 20 and enzyme SEQ. ID No. : 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (75 mL based on settled volume) was added to a filter funnel and washed with water (9 column volumes, 3 x 225 mL) and binding buffer (1 column volume, 75mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0). In a vessel pantothenate kinase (SEQ ID NO. : 20, 6.0 g) lyophilized powder was resuspended in binding buffer (200 mL) and the washed resin was added. The solution was mixed using rotating mixer at 25 °C for 6h. The resin was filtered and washed with binding buffer (6 column volumes, 6 x 225 mL) and BIS-TRIS buffer (8 column volumes, 600 mL; 50 mM, pH 6.2).
Reaction procedure:
An aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (574 g, 8.7% wt, 0.430 mol) and water (350 mL) was charged to a jacketed reactor, followed by 1M BIS-TRIS methane buffer pH 6.5 (50 mL) and magnesium chloride (2.033 g, 0.01 mol). ATP (2.37g, 0.0043 mol, 0.01 equivalent) and diammonium acetyl phosphate (101 g, 89%, 0.530 mmol, 1.2 eq) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using 5 M KOH.
Resin with immobilized pantothenate kinase SEQ. ID No. : 20 (25 mL) and evolved acetate kinase SEQ. ID No. : 21 (0.15 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which the pH dropped to 5.5. Quantitative conversion of 2-ethynyl-propane- I,2,3-triol (3) to (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) was obtained as judged by ¾ and 31P NMR. ¾ NMR (D20, 500 MHz) d 3.89 (m, 2H), 3.72 (d, J= 11.6 Hz, 1 H), 3.65 (d, J =
I I .6 Hz, 1H), 2.93 (s, 1H).
Alternate Preparations of (i?V2-ethvnyl-glvceraldehvde 3-phosphate 15):
Method HI: Immobilized galactose oxidases SEP ID No.: 16
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 16 g of washed resin. In a vessel evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the glycosylation reaction.
Reaction procedure:
The resin with immobilized galactose oxidase SEQ ID NO.: 16 (3.0 g) was added to a solution of S)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (f?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the(f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used
directly in the glycosylation reaction. iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H2: Immobilized galactose oxidases SEP ID No.: 17
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give l6g of washed resin. In a vessel, evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS methane buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the reaction.
Reaction procedure:
The resin with immobilized evolved galactose oxidase SEQ ID NO.: 17 (3.0 g) was added to a solution of (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was used directly in the glycosylation reaction. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H3: Immobilized galactose oxidases SEQ ID No.: 18
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 18, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution ((9), 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 90% conversion and the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H),
4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (MΉ): 193.1; found 193.0.
Method H4: Immobilized galactose oxidases SEP ID No.: 19
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 19, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution (9, 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 100% conversion and (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was obtained in >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
PATENT
CA 2502109
WO 2017053216
US 20200010834
US 20200010868
PAPER
Organic letters (2017), 19(4), 926-929.
Organic Letters (2017), 19(4), 926-929.
Journal of medicinal chemistry (2018), 61(20), 9218-9228.
Bioscience, Biotechnology, and Biochemistry (2020), 84(2), 217-227.
PAPER
Organic letters (2011), 13(19), 5264-6.
A concise enantioselective total synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), an extremely potent anti-HIV agent, has been accomplished from (R)-glyceraldehyde acetonide in 18% overall yield by a 12-step sequence involving a highly diastereoselective ethynylation of an α-alkoxy ketone intermediate.

Processes for preparing islatravir and its analogs comprising the reaction of a substituted tetrahydrofuran compound with purine nucleoside phosphorylase and a nucleobase, followed by stereochemical synthesis, glycosylation, reduction, oxidation and isolation are claimed. Also claimed are novel intermediates of islatravir and processes for their preparation and their use for the preparation of islatravir.
(2R,3S,5R)-5-(6-Amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)- tetrahydrofuran-3-ol (1). To a stirred solution of 16 (66.2 mg, 0.115 mmol) in MeOH/CH2Cl2 (2:1, 1.5 mL) was added NH4F (85.1 mg, 2.30 mmol) at room temperature. After 16 h, MeOH (0.5 mL) was added, and the resulting mixture was stirred for an additional 27 h. To the mixture was added 10% NaOH in MeOH (1.5 mL) to adjust the pH of the mixture to ca. 10. After 10 min, Dowex 50W×8 (200– 400 mesh (H)) was added until the pH of the mixture reached ca. 4. To the resulting mixture was added CaCO3 (259 mg, 2.59 mmol), and the mixture was stirred for 30 min. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to give 29.3 mg (87%) of 1. Mp: 220.0–221.4 °C (dec.); [α] 25 D +12.4 (c 0.97, MeOH); IR: νmax 3315 (br m), 3179 (br m), 1690 (vs), 1356 (vs); 1 H NMR (600 MHz, DMSO-d6): δ 2.43 (1H, ddd, J = 13.2, 7.3, 7.3 Hz), 2.70 (1H, ddd, J = 13.2, 6.8, 5.1 Hz), 3.52 (1H, s), 3.54 (1H, dd, J = 11.7, 6.4 Hz), 3.65 (1H, dd, J = 11.7, 5.0 Hz), 4.57 (1H, m), 5.32 (1H, m), 5.60 (1H, m), 6.24 (1H, dd, J = 7.2, 5.1 Hz), 7.82 (1H, br s), 7.92 (1H, br s), 8.31 (1H, s); 13C NMR (150 MHz): δ 38.3, 64.4, 70.3, 79.2, 81.7, 82.2, 85.4, 117.6, 140.0, 150.4 (d, JCF = 20.7 Hz), 157.8 (d, JCF = 21.2 Hz), 158.8 (d, JCF = 203.4 Hz); HRMS (FAB): m/z calcd for C12H13FN5 O3, 294.1002; found, 294.1000 ([M+H]+ ).
https://pubs.acs.org/doi/suppl/10.1021/ol202116k/suppl_file/ol202116k_si_001.pdf


PAPER
Organic Letters (2011), 13(19), 5264-5266.
PAPER
Bioscience, biotechnology, and biochemistry (2012), 76(6), 1219-25.
https://www.tandfonline.com/doi/abs/10.1271/bbb.120134
EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine), a nucleoside reverse transcriptase inhibitor with extremely potent anti-HIV activity, was concisely synthesized from (R)-glyceraldehyde acetonide in an 18% overall yield by a 12-step sequence involving highly diastereoselective ethynylation of an α-alkoxy ketone intermediate. The present synthesis is superior, both in overall yield and in the number of steps, to the previous one which required 18 steps from an expensive starting material and resulted in a modest overall yield of 2.5%.
PAPER
Bioscience, Biotechnology, and Biochemistry (2012), 76(6), 1219-1225.
Organic letters (2015), 17(4), 828-31.
Organic Letters (2015), 17(4), 828-831.
PAPER
https://cen.acs.org/pharmaceuticals/process-chemistry/Scientists-made-HIV-drug-using/97/web/2019/12

Making small-molecule drugs usually goes something like this: set up a reaction, purify the intermediate, change a solvent, and repeat, repeat, repeat to get the final product. But there’s a lot of waste involved, which is why chemists stress the environmental benefits of an alternate approach: biocatalysis. Engineering enzymes to make reactions happen saves a lot of materials, minimizes chemical and hazardous waste, and even uses less plasticware and glassware. And not having to isolate intermediates saves time.
Some pharmaceutical companies are investigating biocatalysis at different points in their drug development pipelines, but mostly at one or two steps into the making of a small molecule. Scientists at Merck & Co. have taken this further—they are reporting an entire drug synthesis using a chain of nine enzymes, five of which had been engineered, to produce an experimental HIV drug at high yield in just a few steps (Science 2019, DOI: 10.1126/science.aay8484).
This biocatalytic cascade is turning heads. For the most part, scientists aren’t using biocatalysis to manufacture a compound so much as to develop it, says Princeton University chemist Todd Hyster. The Merck process stitches together nine enzymes to get good yields of the final product, which Hyster says is no small feat.
“I was blown away,” Hyster says of the first time he saw Merck scientists talk about this work. “It’s something that was very complicated.”
Mark Huffman, a chemist who led the work at Merck with Anna Fryszkowska, says they turned to biocatalysis in order to overcome a couple of key hurdles in synthesizing some molecules. One is stereochemistry. Islatravir is a nucleoside that blocks the HIV enzyme reverse transciptase and traditionally, in medicinal chemistry, it’s been hard to get the stereochemistry of nucleosides right, Huffman says. But this is something enzymes are designed by nature to do. The other is preventing unwanted side reactions. A number of steps in the traditional chemical synthesis of islatravir put the compound’s functional groups at risk of being lopped off, so they must be protected. Huffman says enzymes are specific in the types of reactions they catalyze, so there’s little to no risk of an unwanted side reaction.
On top of that, Huffman says, they are doing these reactions at neutral pH, in aqueous solvents, and at room temperature, which cuts down on electricity and the need for multiple bioreactors running under different conditions. Islatravir normally takes between 12 and 18 steps to make. With biocatalysis, the team has cut this down to three.
“You don’t have rigorous equipment requirements,” he says. “You’re usually running [these reactions] under much milder conditions.”
To run the cascade, the team started with 2-ethynylglycerol, and added a mixture of three enzymes to run one group of reactions. They then added more enzymes to drive a second set of reactions. Then, they remove the enzymes from the solution, which are immobilized and easy to filter out, and use four more enzymes to drive the final reactions that lead to islatravir. There are no intermediate purification steps. The overall yield is about 51% using biocatalysis, compared to yields of 7% and 15% using two more traditional syntheses.
To make their biocatalysts, the team surveyed natural enzymes, mostly from microbes, that interacted with the different intermediates in islatravir production. One of the reasons why Huffman says islatravir is an ideal small molecule to produce using biocatalysis is that most organisms have to make and break down nucleosides, so there are several natural enzymes found across multiple species. This gave the team a lot of starting material from which to alter amino acids and build the enzymes they needed to do their syntheses. By making adjustments to active sites and other areas of the enzymes, the team built five of the nine enzymes needed to make islatravir biochemically.
Huffman says that while islatravir is a good molecule to show that scientists can build large biocatalytic cascades, Merck is also looking at biocatalysis to make other small molecules and biologic drugs.
Alison Narayan, a biocatalysis chemist at the University of Michigan, calls Merck “bold” for putting the time, money, and people behind this change in production—it takes a lot of resources to try an entire synthesis via biocatalysis. And, she says, they’ve succeeded spectacularly. “It literally took my breath away,” Narayan says of her first exposure to this project in 2018. “I think it’s a huge accomplishment.”
She says that Merck’s islatravir work shows that industry is starting to appreciate what biocatalysis can do for their drug pipelines and their financial bottom lines. Alongside Merck, companies like GlaxoSmithKline and Pfizer are also exploring biocatalysis at different points in drug development and manufacturing.
“It’s an important proof of concept,” Narayan says. “This is a practical way to build molecules, and this will be the way that people will build molecules when you take into consideration efficiency, green-ness, and constructing an effective synthesis. Biocatalysis has a lot to offer.”
PAPER
Biocatalytic cascades go viral
Natural biosynthesis assembles a vast array of complex natural products starting from a limited set of building blocks, under physiological conditions, and in the presence of numerous other biomolecules. Organisms rely on the extraordinary selectivity of enzymes and their ability to operate under similar reaction conditions, meaning that these catalysts are perfectly adapted to mediate cascade reactions. In these multistep processes, the product of one biocatalytic step becomes the substrate for the next transformation (Display footnote number:1-3). On page 1255 of this issue, Huffman et al. (Display footnote number:4) report the development of an impressive nine-enzyme biocatalytic cascade for the synthesis of the investigational drug islatravir for the treatment of human HIV.
This study represents a partnership between scientists from Merck and Codexis. These two companies have a history of successfully collaborating to develop biocatalysts for the synthesis of important pharmaceuticals. Almost a decade ago, they developed a chemoenzymatic route for the synthesis of the type 2 diabetes drug sitagliptin (Januvia), relying on a key enzyme-catalyzed transamination with a highly engineered (R)-selective transaminase (Display footnote number:5). The work was considered a landmark example of directed evolution and functioned to highlight the potential application of biocatalysis to revolutionize industrial chemical processes.
The cascade for synthesizing islatravir was inspired by the bacterial nucleoside salvage pathway, which recycles precious nucleosides by using three key enzymes: a purine nucleoside phosphorylase (PNP), a phosphopentomutase (PPM), and a deoxyribose-5-phosphate aldolase (DERA) (see the figure). However, to achieve the synthesis of the target molecule, Huffman et al. required the natural nucleoside degradative cascade to run in reverse. The reversible nature of enzymes is central to the design of this cascade and is one of the important features that sets biocatalysts apart from the majority of traditional chemical catalysts.
The success of the cascade developed by the team also relied on all three enzymes accepting non-natural substrates bearing a fully substituted carbon at the C-4 position of the 2-deoxyribose ring. The authors reconstructed the reverse nucleoside salvage pathway from a PNP and PPM found in Escherichia coli and a DERA from Shewanella halifaxensis. The native E. coli enzymes required engineering to improve their activity. The DERA displayed existing high activity and stereoselectivity for the formation of the desired sugar phosphate enantiomer, but it required engineering to improve its ability to operate at high substrate concentration.
One of the many advantages of performing biocatalytic cascade reactions is the effective displacement of unfavorable reaction equilibria that can be achieved through product removal. However, despite performing the PNP and PPM steps in tandem, the reaction proceeded with poor conversion, and the inorganic phosphate by-product inhibits the enzymes. An elegant solution to these issues was the inclusion of an auxiliary sucrose phosphorylase, along with its sugar substrate, which removed free phosphate and effectively displaced the reaction equilibrium toward product formation.
Having assembled enzymes for the three key steps in the cascade, Huffman et al. sought to develop a biocatalytic route for the synthesis of the DERA substrate 2-ethynylglyceraldehyde 3-phosphate. Extensive screening of a broad range of kinases resulted in the identification of pantothenate kinase (PanK) from E. coli, which displayed low levels of activity (∼1% conversion) toward the (R)-enantiomer of the target aldehyde. Despite the modest initial activity, directed evolution was successfully used to substantially improve the productivity and stability of this enzyme. Finally, after 12 rounds of evolution, the authors reversed the enantioselectivity and improved the activity, stability, and expression of a galactose oxidase variant for the desymmetrization of the starting substrate, 2-ethynylglycerol.
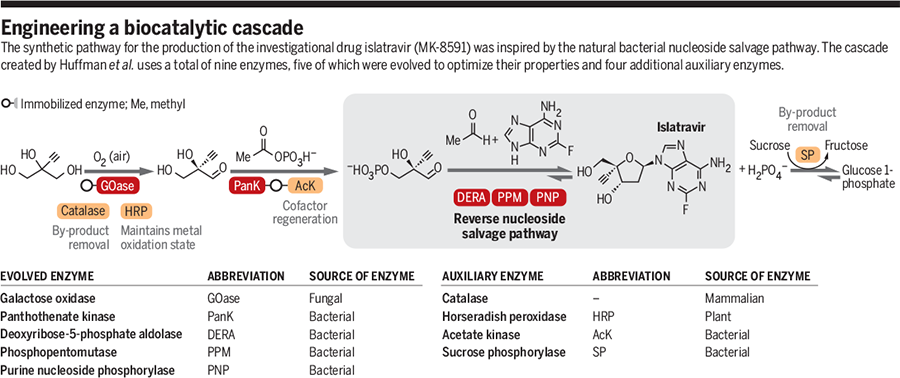
Viewable Image – engineering a biocatalytic cascade
Image Caption
GRAPHIC: A. KITTERMAN/SCIENCE
Advancements in protein engineering, rapid gene sequencing, and the availability of low-cost DNA synthesis have made it possible to alter the properties of enzymes and fine-tune them for biocatalytic applications (Display footnote number:6-8). The work by Huffman et al. is a milestone in cascade design, largely because of the number of biocatalysts operating in tandem and the engineering feat required to optimize five of the nine enzymes involved in the synthesis. It also highlights how biosynthetic or degradative pathways can be a source of inspiration for the design of efficient biocatalytic cascades and how sequences can be reconstituted using enzymes recruited from multiple sources—in this case, of bacterial, fungal, plant, and mammalian origin. The diverse role that biocatalysts can play is also exemplified in this work, where five engineered enzymes are directly involved in the synthesis of the target molecule, and four additional enzymes function to recycle coenzyme, remove inhibitory by-products, and maintain the correct oxidation state of the copper cofactor.
Previous approaches reported for the synthesis of islatravir relied on multistep syntheses and require protecting group manipulations and intermediate purification steps (Display footnote number:9, 10). The incorporation of a key biocatalytic step or steps has the potential to revolutionize synthetic design strategies by making possible transformations that are not accessible using solely chemical approaches (Display footnote number:11, 12). The application of enzymes in industry and the development of chemoenzymatic routes to complex molecules is now well established. However, multistep syntheses exclusively comprising biocatalytic transformations are rare (Display footnote number:13), and this contribution sets a new standard for the synthesis of complex molecules with enzymatic cascades.
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. Email: elaine.oreilly@ucd.ie.
REFERENCES AND NOTES
ACKNOWLEDGMENTS
J.R. acknowledges the School of Chemistry, University College Dublin, for support.
References
- ^ Kawamoto, A; Kodama, E; Sarafianos, SG; Sakagami, Y; Kohgo, S; Kitano, K; Ashida, N; Iwai, Y; Hayakawa, H; Nakata, H; Mitsuya, H; Arnold, E; Matsuoka, M (2008). “2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants”. The International Journal of Biochemistry & Cell Biology. 40 (11): 2410–20. doi:10.1016/j.biocel.2008.04.007. PMID 18487070.
- ^ Roy M. Gulick (2018). “Investigational Antiretroviral Drugs: What is Coming Down the Pipeline”. Top Antivir Med. 25 (4): 127–132. PMC 5935216. PMID 29689540.
- ^ “Someday, an Arm Implant May Prevent H.I.V. Infection for a Year”. New York Times. July 23, 2019.
- ^ “Merck Presents Early Evidence on Extended Delivery of Investigational Anti-HIV-1 Agent Islatravir (MK-8591) via Subdermal Implant”(Press release). July 23, 2019.
- ^ Jump up to:a b Michailidis, Eleftherios; Huber, Andrew D.; Ryan, Emily M.; Ong, Yee T.; Leslie, Maxwell D.; Matzek, Kayla B.; Singh, Kamalendra; Marchand, Bruno; Hagedorn, Ariel N.; Kirby, Karen A.; Rohan, Lisa C.; Kodama, Eiichi N.; Mitsuya, Hiroaki; Parniak, Michael A.; Sarafianos, Stefan G. (2014). “4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) Inhibits HIV-1 Reverse Transcriptase with Multiple Mechanisms”. Journal of Biological Chemistry. 289 (35): 24533–48. doi:10.1074/jbc.M114.562694. PMC 4148878. PMID 24970894.
- ^ Grobler, Jay (February 22–25, 2016). Long-Acting Oral and Parenteral Dosing of MK-8591 for HIV Treatment or Prophylaxis. Boston, Massachusetts. Conference on Retroviruses and Opportunistic Infections. 98.
- ^ Stoddart, Cheryl A.; Galkina, Sofiya A.; Joshi, Pheroze; Kosikova, Galina; Moreno, Mary E.; Rivera, Jose M.; Sloan, Barbara; Reeve, Aaron B.; Sarafianos, Stefan G.; Murphey-Corb, Michael; Parniak, Michael A. (2015). “Oral Administration of the Nucleoside EFdA (4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque”. Antimicrobial Agents and Chemotherapy. 59 (7): 4190–8. doi:10.1128/AAC.05036-14. PMC 4468726. PMID 25941222.
- ^ Bruno Marchand. “The Crystal Structure of EFdA‐Resistant HIV‐1 Reverse Transcriptase Reveals Structural Changes in the Polymerase Active Site” (PDF).
|
|
| Names | |
|---|---|
| IUPAC name
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
|
|
| Other names
EFdA; MK-8591
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C12H12FN5O3 | |
| Molar mass | 293.258 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////////Islatravir, MK-8591, EFdA, PHASE 2, HIV-1 , HIV-2,
C#CC1(C(CC(O1)N2C=NC3=C(N=C(N=C32)F)N)O)CO