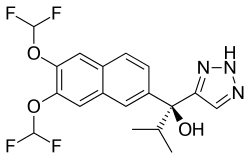
SEVITERONEL
CAS Registry Number 1610537-15-9
Molecular formulaC18 H17 F4 N3 O3, MW 399.34
1H-1,2,3-Triazole-5-methanol, α-[6,7-bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-, (αS)-
(αS)-α-[6,7-Bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-1H-1,2,3-triazole-5-methanol
8S5OIN36X4
- Mechanism of ActionAndrogen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- WHO ATC codeL01 (Antineoplastic Agents)L01X-X (Other antineoplastic agents)
- EPhMRA codeL1 (Antineoplastics)L1X9 (All other antineoplastics)
1H-1,2,3-Triazole-5-methanol, alpha-(6,7-bis(difluoromethoxy)-2-naphthalenyl)-alpha-(1-methylethyl)-, (alphaS)-
Seviteronel (developmental codes VT-464 and, formerly, INO-464) is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer.[1] It is a nonsteroidalCYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body.[1] As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer.[1] In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer.[1][2] In April 2017, seviteronel received fast-track designation for breast cancer as well.[1]
- Originator Viamet Pharmaceuticals
- Developer Innocrin Pharmaceuticals
- Clas sAntiandrogens; Antineoplastics; Fluorine compounds; Naphthalenes; Propanols; Small molecules; Triazoles
- Mechanism of Action Androgen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- Phase II Breast cancer; Prostate cancer; Solid tumours
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial in Prostate Cancer (Second-line therapy or greater, Hormone refractory) in the US (NCT02445976)
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial for Prostate Cancer (Hormone refractory) in the US, UK, Switzerland and Greece (NCT02012920)
- 31 Jan 2019 Innocrin Pharmaceuticals completes the phase I/II CLARITY-01 trial for Breast cancer (Late stage disease) in USA (NCT02580448)
- CYP-17 useful for treating fungal infections, prostate cancer, and polycystic ovary syndrome, assigned to Viamet Pharmaceuticals Inc , naming Hoekstra and Rafferty. Innocrin Pharmaceuticals , a spin-out of Viamet is developing oral seviteronel, the lead dual selective inhibitors of the 17,20-lyase activity of P450c17 (CYP17) and androgen receptor antagonist, which also includes VT-478 and VT-489, developed using the company’s Metallophile technology, for treating castration-resistant prostate cancer (CRPC) in men, breast cancer and androgen (AR) related cancers.
Pharmacology
Pharmacodynamics
Seviteronel is a nonsteroidal antiandrogen, acting specifically as an androgen synthesis inhibitor via inhibition of the enzyme CYP17A1, for the treatment of castration-resistant prostate cancer.[3][4][5][6][7][8] It has approximately 10-fold selectivity for the inhibition of 17,20-lyase (IC50 = 69 nM) over 17α-hydroxylase (IC50 = 670 nM), which results in less interference with corticosteroid production relative to the approved CYP17A1 inhibitor abiraterone acetate (which must be administered in combination with prednisone to avoid glucocorticoid deficiency and mineralocorticoid excess due to 17α-hydroxylase inhibition) and hence may be administerable without a concomitant exogenous glucocorticoid.[4][5][6][7][8] Seviteronel is 58-fold more selective for inhibition of 17,20-lyase than abiraterone (the active metabolite of abiraterone acetate), which has IC50 values for inhibition of 17,20-lyase and 17α-hydroxylase of 15 nM and 2.5 nM, respectively.[7] In addition, in in vitro models, seviteronel appears to possess greater efficacy as an antiandrogen relative to abiraterone.[6] Similarly to abiraterone acetate, seviteronel has also been found to act to some extent as an antagonist of the androgen receptor.[6]
Society and culture
Generic names
Seviteronel is the generic name of the drug and its INN.[9]
PATENT
WO2012064943
PATENT
WO-2019113312
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019113312&redirectedID=true
The present invention relates to a process for preparing compound 1 that is useful as an anticancer agent. In particular, the invention seeks to provide a new methodology for preparing compound 1 and substituted derivatives thereof.
Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-( 1,2, 4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes.
One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently-available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull. 1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable
Preparation of Compound 4:
de
Acetone (850 L), 2,3-dihydroxynaphthalene (85.00 kg, 530.7 moles), and potassium carbonate (219.3 kg, 1,586.7 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. Dimethyl sulfate (200.6 kg, 2131.09) was added to the stirred reaction at a rate that maintains the internal temperature of the exothermic reaction below 60 °C. This addition typically requires about 3 hours. At the end of the dimethyl sulfate addition, the reaction is continued to allow to stir while maintaining the internal temperature at 50 – 60 °C. After about 3 hours, the reaction was analyzed by HPLC. The reaction was concentrated by atmospheric pressure distillation of acetone. The distillation was continued until 340 – 425 L of distillate was collected. This represents 40 – 50 % of the initial charge of acetone. At the end of the distillation, the reaction mass is present as a thick suspension. While maintaining the internal temperature below 60 °C, the reactor contents were slowly diluted with water (850 L). When the addition is complete, the reaction was cooled to an internal temperature of 25 – 35 °C and stirring was continued for 1 – 2 hours after the designated internal temperature was reached. Compound 2 was isolated by filtration and the cake was washed with water (at least 3 X 85 L). Compound 2 was dried at 40 – 45 °C and full vacuum until the water content by Karl Fisher titration is found to be NMT 2.0 %. Typically, greater than 90 kg of dry product is obtained with an assay of >99.5% AUC by HPLC.
Dichloromethane (with a water content by Karl Fisher Titration of NMT 0.50%) (928 L) and 2,3-dimethoxynaphthalene (2, 116.00 kg, 616.3 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. The reactor contents were cooled to an internal temperature of -5 to 0 °C. Aluminum chloride (164.72 kg, 1235.3 moles, 2.00 molar equivalents) was carefully added in portions to the reaction, while maintaining the internal temperature at -5 to +5 °C. This addition typically requires 5 – 6 hours. At the end of the addition, the reactor contents were cooled to an internal temperature of -15 to -5 °C. Isobutyryl chloride (102.08 kg, 958.05 moles, 1.55 molar equivalents) was slowly added to the reaction while maintaining the internal temperature at -15 to -5 °C. The addition typically requires about 3 hours. At the end of the isobutyryl chloride addition, the reaction was warmed to an internal temperature of 20 – 35 °C. When the temperature was reached, these conditions were maintained for 2 – 3 hours until the IPC indicated a level of residual starting material of NMT 2.0 % AUC by HPLC. The reactor contents were then cooled to 0 – 5 °C. The reaction was quenched by adding the reaction to a precooled (0 – 5 °C) 3M aqueous solution of hydrochloric hcid (Water, 754 L: cone. HC1, 406 L). The mixture was vigorously stirred for 15 – 20 minutes then the layers were allowed to settle. The lower, dichloromethane, product-containing layer was washed sequentially with 10 % aqueous sodium bicarbonate (1044 L), water (1160 L), then 10 % aqueous sodium chloride (1044 L). The reaction was concentrated by distillation under full vacuum and at an internal temperature of NMT 40 °C. The reaction concentrate was cooled to 20 – 35 °C and diluted with hexanes (812 L). The resultant slurry was warmed to 45 – 50 °C and these conditions were maintained for 1 – 2 hours. The reactor contents were cooled to 20 – 35 °C for 1 – 2 hours. Compound 3 was isolated by filtration. The cake was washed with fresh hexanes (232 L) twice, the filter was cooled, and the cake was washed an additional two times with hexanes. Compound 3 was dried under full vacuum at a jacket temperature of 45 °C. Typically, about 95 kg of dry product was isolated with a product purity of >90% by HPLC.
Acetic acid (212.5 L L) and l-(6,7-dimethoxynaphthalene-2-yl)-2-methylpropane-l- one (42.5 kg, 164.5 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 25 – 45 °C. Concentrated hydrochloric acid (425.0 L) was added carefully to the stirring reactor contents while maintaining reactor contents at an internal temperature of 25 – 45 °C. When the addition was complete, the internal temperature of the reaction was raised to 100 – 105 °C. Note that the reaction is a heterogeneous mixture. The reaction was stirred under these conditions for 6 – 8 hours. The reaction was cooled to 85 – 90 °C to which was carefully added a fresh portion of hydrochloric acid (127.5 L). The reaction was warmed to 100 – 105 °C and stirred for another 6 – 8 hours. The reaction was cooled to 85 – 90 °C. The reaction was cooled further to 70 – 80 °C. Water (212.5 L) was added to the well stirred reaction and the reactor contents were cooled to an internal temperature of 35 – 45 °C and stirred for 3 – 4 hours. Compound 4 was collected by filtration. The wet cake was washed with water (212.5 L). The wet cake was added to a clean reactor with a 5% aqueous sodium bicarbonate solution and stirred at an internal temperature of 35 – 45 °C for 1 – 2 hours.
Compound 4 was collected by filtration and washed with water (212.5 L). Compound 4 was dried under full vacuum and a temperature of < 50 °C until the water content of the dried material was found to be NMT 5.0% by Karl Fisher Titration. The yield is typically >31 kg with a purity >99.5 %.
Preparation of Compound 5:
The following difluoromethylation conditions listed in Table 1 were investigated:
Preparation 1:
The reaction flask was dried under an argon flow at 120 °C. (lS,2R)-l-Phenyl-2-(l- pyrrolidinyl)propan-l-ol (ligand 45) (196.6 g, 0.96 mol, 2.2 eq.) was added into the flask and then toluene (195 mL) was added. The solution was cooled to <12 °C. A solution of diethyl zinc (716.4 g, 0.87 mol, 15 wt%, 2 eq.) in toluene was added through a septum over 30 min at 0-10 °C. Further, a solution of ((Trimethylsilyl)ethynyl)-magnesium bromide in THF (1.81 kg; 0.87 mol, 9.7 wt%, 2 eq.) was added over 30 min at 0-10 °C. Finally, trifluoroethanol (87.0 g; 0.87 mol; 2 eq.) was added over 10 min at 0-10 °C. The reaction solution was stirred at 10-12 °C for 3 h. Compound 5 (143.4 g; 0.434 mol; 1 eq.) was added (as a solid) at room
temperature. The reaction mixture was stirred at room temperature for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed with aqueous HC1 (3600 mL; 7.5 wt%) within 20 min. The temperature of the mixture was kept below 25 °C. Toluene (1250 mL) was added and the mixture was stirred at room temperature for 5 min. The aqueous phase was separated and stored for the recycling of ligand 45. The organic phases were washed with water (638 mL) and concentrated via distillation under reduced pressure (50 mbar). The residue (approx. 184 g) was treated with heptane (200 mL), which was removed
via distillation. The residue was dissolved in heptane (2050 mL) at 50 °C. The mixture was cooled to room temperature and subsequently to -8 °C within 2 hours. The obtained suspension was stirred at -8 °C for 1 h. Crystallized compound 5 (20.0 g; 14%) was isolated via filtration, washed twice with cold (0 °C) heptane (2×20 mL) and dried under vacuum at 50 °C for 12 hours. The combined heptane phases were concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (yield: 83.0%). The solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Preparation 2:
(7S,2R)-l-Phenyl-2-(l-pyrrolidinyl)propan-l-ol (ligand 45) (13.0 kg, 63.3 mol, 2.2 eq.) was charged into the reactor and toluene (60 L) was added. The solution was cooled to < 12 °C. A solution of diethyl zinc (35.6 kg, 57.3 mol, 20 wt%, 2 eq.) in toluene was added via mass flow controller at 8-16 °C. Further, a solution of ((trimethylsilyl)ethynyl)-magnesium bromide in THF (11.5 kg; 57.3 mol, 9.7 wt%, 2 eq.) was added at 8-16 °C. Finally, trifluoroethanol (5.7 kg; 57.3 mol; 2 eq.) was added over 10 min at 8-16 °C.The reaction solution was stirred at 22-25 °C for 3 h. A solution of compound 5 (9.5 kg; 28.7 mol; 1 eq.) in toluene (20 L) was added at room temperature. The reaction mixture was stirred at 25 °C for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed in aqueous HC1 (225L; 7.5 wt%) within 20 min. The temperature of the mixture should be kept below 25 °C. Toluene (80 L) was added and the mixture was stirred at room temperature for 5 min. The organic phases was washed with water (50 L) and concentrated via distillation under reduced pressure (50 mbar). The residue was treated with heptane (100 L), which was removed via distillation. The residue was dissolved in heptane (100 L) at 50°C, which was removed via distillation. The residue was dissolved in heptane (25 L). Heptane (110 L) was added, the mixture was cooled to room temperature and subsequently to 0-5 °C and seeded with compound 5 (0.15 kg). The obtained suspension was cooled to -8 °C within 1 h and stirred at this temperature for 2 h. Crystallized compound 5 was removed via filtration. The filtrate was concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (calculated 8.8 kg, 71.6%). This solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Recovery of the chiral ligand ( lS,2R)-l-Phenvl-2-
-l-ol from the
Preparation 1:
The above acidic aqueous phase was diluted with toluene (1000 mL) and the mixture was treated with sodium hydroxide (50 wt% solution) to adjust the pH to 12. The mixture was warmed to 50 °C and sodium chloride (100 g) was added. The aqueous phase was separated and washed with toluene (1000 mL). The combined organic phases were washed with water (200 mL). The combined toluene phases were treated with water (1000 mL) and the pH was adjusted to 2 by the addition of a cone. HC1 solution. The aqueous phase was separated and the mixture was treated with sodium hydroxide (50 wt% solution) at 5 °C to adjust the pH to 12. After seeding, the suspension was stirred at 5 °C for 30 min. The solids were isolated, washed with cold (0 °C) water (4×100 mL) and dried under vacuum at 30 °C for 24 hours. Ligand 45 (178.9g; 91%) was obtained as slightly yellow crystalline solid.
HPLC (purity): 99%.
Preparation 2:
The acidic aqueous phase containing ligand 45 (500 L) was diluted with toluene (125 L) and treated with“Kieselgur” (20 L). The mixture was treated with sodium hydroxide (40 L; 50 wt% solution) to adjust the pH to 12 whereas the temperature was kept <55 °C. The suspension was stirred for 15-20 min and filtered to remove all solids. Toluene (80 L) was added and the aqueous phase was separated. The organic phase was treated with water (150 mL) and the pH was adjusted to 1.5-2 by the addition of an aqueous HC1 solution (10 L; 32 wt%). The aqueous phase was separated, toluene (150 L) was added, and the mixture was treated with sodium hydroxide (5 L; 50 wt% solution) at 5 °C to adjust the pH to 12-12.5. The organic phase was separated, washed with water (30 L), and concentrated under reduced
pressure at 50 °C. Approx. 100L of distillate was removed. A sample of the solution of ligand 45 in toluene was analyzed:
The NMR results indicated a 21.6 wt% solution of ligand 45 in toluene which corresponds to a calculated amount of 118.4 kg (83.6%) of ligand 45.
Preparation of Compound 18a
Preparation 1:
A solution of tertiary alcohol 18b (320 g; 48 wt%; 0.36 mol; 1 eq.) in heptane was dissolved in methanol (800 mL). Potassium carbonate (219 g; 1.58 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at room temperature for 3 h. Water (1250 mL) was added and the mixture was treated with a cone. HC1 solution (approx. 130 mL) to adjust the pH to 7.8. The reaction mixture was extracted twice with methyl- /-butyl ether (MTBE; 2×465 mL). The combined MTBE phases were washed with water (155 mL). Water (190 mL) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (50 mbar). The obtained emulsion of compound 18a (yield: 99%) was directly used for the next step.
1H-NMR (600.6 MHz, CDC13) d: 0.87 (d, J = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / =
8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation 2:
The solution of tertiary alcohol 18b (48 wt%; 57.5 mol; 1 eq.) in heptane was dissolved in methanol (128 L). Potassium carbonate (35.0 kg; 253 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at 20-30 °C for 3 h. Water (200 L) was added and the mixture was treated with an aqueous HC1 solution (approx. 25 L; 32 wt%) to adjust the pH to 7.5 – 7.8. The reaction mixture was extracted twice with MTBE
(2×66.6 L). The combined MTBE phases were washed with water (25 L). Water (30 L) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (<80 mbar; 55°C). The residue was dissolved in tert-butanol (25 L). The resulting 18a was cooled to <30°C and used directly in the next step.
^-NMR (600.6 MHz, CDC13) d: 0.87 (d, / = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / = 8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation of Compound 31
Preparation 1:
Benzyl bromide (39.4 g; 0.23 mol; 1 eq.) was dissolved in water (177 mL) and t-BuOH (200 mL). Diisopropylethylamine (DIPEA; 59.4 g; 0.46 mol; 2 eq.) and sodium azide (15.0 g; 0.23 mol; 1 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of compound 18a (82 g; 0.23 mol; 1 eq.) in water (123 mL) was treated with t-BuOH (100 mL) and copper (I) iodide (8.8 g; 46 mmol; 0.2 eq.) was added and the temperature was kept below 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (5.0 g; 76 mmol) and ammonium chloride (7.4 g; 0.14 mol) were added and the reaction mixture was stirred at room temperature for 3 hours. The mixture was diluted with MTBE (800 mL), water (280 mL), and an aqueous ammonia solution (120 g; 25 wt%). Solids were removed by filtration and additional MTBE (200 mL) and brine (200 mL) were added. The aqueous phase was separated and extracted with MTBE (400 mL). The combined organic phases were treated with water (150 mL) and MTBE was distilled off under reduced pressure (100 mbar). The obtained suspension of compound 31 (113 g; 50 wt%) in water (approx. 113 mL) was directly used for the next step.
Ή-NMEI (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 2:
Benzyl bromide (11.0 kg g; 64.4 mol; 1,12 eq.) was dissolved in water (40 L) and t-BuOH (60 L). DIPEA (16.4 kg; 126.5 mol; 2,2 eq.) and sodium azide (4.12 kg; 63.3 mol; 1 eq.) were added. The suspension was stirred 5 min at room temperature. A mixture of compound 18a (20.5 kg; 57.5 mol; 1 eq.) in ieri-butanol (see previous step) was added together with water (5 L) and copper (I) iodide (2.2 kg; 11.5 mol; 0.2 eq.) at a temperature < 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (1.25 kg; 19 mol, 0.33 eq.) and an aqueous solution of ammonium chloride (2.14 kg; 20 wt%; 40 mol; 0.7 eq.) were added and the reaction mixture was stirred at 20-30 °C for 2 hours. The reaction mixture was concentrated under vacuum (<200 mbar, 55 °C). The residue was diluted with MTBE (200 L), water (30 L), and an aqueous ammonia solution (30 kg; 25 wt%). Solids were removed by filtration over a pad of“Kieselgur NF” (2 kg). Brine (50 L) was added for a better phase separation. The aqueous phase was separated and washed with MTBE (200 L). The combined organic phases were washed with an aqueous HC1 solution (1 N, 52 L) and water (50 L). MTBE was distilled off under reduced pressure (<400 mbar, 55°C; distillate min. 230L). The oily residue was dissolved in ethanol (150 L), which was distilled off under reduced pressure (<300 mbar; 55°C; distillate min. 150-155L) and the residue was dissolved in additional ethanol (60 L). To the resulting solution of compound 31 was added water (24 L) and the mixture was warmed to 50-55 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (2 x 12 L). The wet product was dissolved in ethanol (115L) at 60 °C and water (24 L) was added. The mixture was cooled to 40 °C and the crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for additional 2 hours. The solids were isolated and washed (without stirring) with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 was isolated as a white solid, which was used for the next step without drying. 14.0 kg of wet 31 were obtained with a 31 content of 81.6 wt%. Based on the determined content, the calculated amount of pure 31 was 11.4 kg with a yield of 41% over two steps (from 18b).
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 3: Synthesis of compound 31 directly from compound 18b
Benzyl bromide (1.64 g, 9.59 mmol, 1.12 eq) was dissolved in water (2.4 mL) and
MeOH (2.4 mL). K2CO3 (2.38 g, 17.2 mmol, 2.00 eq), sodium ascorbate (0.34 g, 1.72 mmol, 0.20 eq) and finally sodium azide (0.62 g, 9.40 mmol, 1.10 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of 18b (3.08 g; 8.64 mmol, 1.00 eq) in water (2.5 mL) and MeOH (2.5 mL) and the resulting mixture was stirred for 10 min.
CuS04 (0.21 g, 1.30 mmol, 0.15 eq) were added (slightly exothermic reaction). The reaction mixture was stirred for 19 h and the conversion was determined by HPLC (conv. 100%, purity of compound 31 by HPLC: 83 area%). To the yellow-green suspension was added zinc powder (0.24 g, 4.13 mmol, 0.43 eq) and ammonium chloride (0.34 g, 6.36 mmol, 0.74 eq) were added and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure (150 mbar, 50 °C). The mixture was diluted with MTBE (40 mL), water (15 mL), and an aqueous ammonia solution (6.5 mL). Solids were removed by filtration and brine (5.5 mL) was added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were treated with water (10 mL) and the pH was adjusted to a pH of 1 by addition of cone. HC1. After phase separation, the organic layer was washed with water (10 mL). MTBE was distilled off under reduced pressure (100 mbar, 50°C) to give the crude compound 31 as an oil. Water (2.5 mL) and EtOH (30 mL) were added and the mixture was warmed to 50 °C. After cooling to 30 °C, the mixture was seeded with compound 31 and compound 31 started to precipitate. The mixture was kept for 1 h at 30 °C, then cooled to 0 °C over 2 h and kept at 0 °C for 2 h. The resulting product, 31, was collected by filtration and the filter cake was washed with small portions of EtOH/water (1:1). After drying, the product (2.97 g) was obtained as a pale yellow, crystalline solid with an HPLC purity of 79 area% and a NMR content of ca. 70 wt%.
Recrystallization of
31
Preparation 1:
To a suspension of compound 31 (96 g; 0.196 mol; 50 wt%) in water (96 mL) was added ethanol (480 mL) and the mixture was warmed to 50 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 40 mL). The wet product was dissolved in ethanol (280 mL) at 60 °C and water (56 mL) was added. The mixture was cooled to 40 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 28 mL). Pure, wet compound 31 (46.8 g on dried basis; 49 % over 2 steps) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.5%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation 2:
14 kg of ethanol-wet 31 (content 81.6 wt%, calculated 11.4 kg, 23.7 mol) were suspended in ethanol (46 L) and the mixture was warmed to 50-55 °C, forming a homogenous solution at this temperature. Water (9 L) was added at 50-55 °C and the mixture was cooled to 40-45 °C. After the crystallization had started, the suspension was stirred at 40-45 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 (14.5 kg) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.8%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation of Azidomethyl Pivalate Protected Triazole (6) from Compound 18a
1
Azidomethyl pivalate (1.42 g, 9.00 mmol, 1.05 eq) was suspended in water (6.0 mL) and t-BuOH (7.2 mL) and the suspension was stirred for 5 min. Compound 18a (theor. 3.08 g, 8.64 mmol, 1.00 eq), sodium ascorbate (0.48 g, 2.4 mmol, 0.30 eq), and CuS04 (0.08 g, 0.40 mmol, 0.05 eq.) were added. The reaction mixture was stirred for 19 h and conversion was determined by HPLC (conv. 98%, purity of the product by HPLC: 81 area%). To the green suspension was added MTBE (20 mL), water (10 mL), and an aqueous ammonia solution (2 g). A biphasic turbid mixture was formed. To improve phase separation, additional MTBE (20 mL) and water (10 mL) were added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give the crude product as a brown oil that solidified upon standing. HPLC purity: ca. 65 area%; NMR content of ca. 73 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Azidomethyl Pivalate Protected Triazole (6) from 18b
In a reaction flask, sodium ascorbate (277 mg, 1.4 mmol, 1.20 eq) and CuS04 (37 mg, 0.23 mmol, 0.20 eq.) were suspended in MeOH (11 mL). Azidomethyl pivalate (183 mg, 1.16 mmol, 1.00 eq) and 18b (183 mg, 1.16 mmol, 1.00 eq) were added and the mixture was warmed to 60 °C. The reaction mixture was stirred for 19 h and worked up. To the green suspension was added an aq NH4Cl solution (2 mL) and zinc powder, and the mixture was stirred for 2 h. MTBE (2 mL) was added and the aqueous phase was separated and extracted with MTBE (2 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give 6 as a brown oil that solidified upon standing. HPLC purity: ca. 81 area%; NMR content of ca. 57 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Compound 1
Preparation 1:
Compound 31 (26 g; 53 mmol; 1 eq.) was dissolved in ethanol (260 mL) and Noblyst Pl 155 (2.2 g; 10 % Pd; 54 wt% water) was added. The autoclave was flushed with nitrogen and hydrogen (5 bar) was added. The reaction mixture was stirred at room temperature for 32 hours. The reaction mixture was treated with charcoal (2 g), stirred for 15 min, and the charcoal was filtered off. The filtrate was concentrated via distillation and the residue (approximately 42 g) was diluted with heptane (200 mL). The mixture was heated to reflux to
obtain a clear solution. The solution was cooled to room temperature within 1 h and the resulting suspension was cooled to 0 °C and stirred for 2 hours at 0 °C. The solids were isolated via filtration and washed with heptane/ethanol (10:1; v/v; 3×10 mL). Compound 1 (18.0 g; 85 %) was dried under vacuum at 60 °C for 24 hours and obtained as a white, crystalline solid.
1H-NMR (600 MHz) d: 0.80 (d, J = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 2:
Compound 31 (26.5 kg; 53.5 mol; 1 eq.) was dissolved in ethanol (265 L) and Pd/C (2.0 kg; 10 % Pd; 54 wt% water) was added. The reactor was flushed with nitrogen, and hydrogen (4.5 bar) was added. The reaction mixture was stirred at 28-32 °C until the reaction was complete. The reaction mixture was treated with charcoal (1.3 kg) at a temperature of <
33 °C, stirred for 10 min, and the charcoal was filtered off, and the filter was washed with ethanol (10 L).The filtrates from two reactions were combined and concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 480 L). The residue (approx. 50-60 L) was diluted with isopropylacetate (250 L). The mixture was again concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 240-245 L). The residue (approx. 60-70 L) was cooled to 35-40 °C and isopropylacetate (125 L) and heptane (540 L) were added. The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). Wet 1 was dried under vacuum at 60 °C and was obtained as a white, crystalline solid (35.4 kg, 81.9%).
1H-NMR (600 MHz) d: 0.80 (d, / = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 3: Preparation of Compound 1 from Compound 6
At room temperature, 6 (3.00 g, 5.84 mmol) was dissolved in MeOH (19.8 mL). NaOH (1.0 M, 19.8 mL) was added in one portion and the reaction mixture was stirred for 1 h at room temperature. The reaction progress was monitored by HPLC, which showed 98% conversion after 1 h. Aq. HC1 (19.8 mL) was added and the mixture was diluted with water (120 mL) and MTBE (60 mL), resulting in a clear biphasic solution. After phase separation, the organic phase was washed with aq NaHC03 (20 mL). The organic layer was concentrated under high vacuum (25 mbar, 45 °C) to yield 2.77 g of 1 as a greenish oil. The identity was confirmed by comparison of HPLC retention time with an authentic sample of 1 as well as by 1H NMR.
Recrystallization of Compound 1
Wet 1 (40 kg; isopropylacetate/heptane wet) was treated with isopropylacetate (110 L) and heptane (440 L). The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with
heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). A sample was taken for analysis
(criterion: a) purity; NLT 99.0 A% by HPLC; b) single impurities, NMT 0.15 A% by HPLC; c) enantiomer VT-463, NMT 1.0 A% by HPLC). Wet 1 was dried under vacuum at 60 °C for not less than 12 h. A sample was taken for analysis: criterion: a) LOD; NMT 0.5 wt% by gravimetry; b) residual toluene, NMT 890 ppm by HS-GC. 1 was obtained as a white, crystalline solid (28.5 kg, 66.7% from 31).
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(11), 2444-2447.
https://www.sciencedirect.com/science/article/pii/S0960894X14003606

PATENT
WO 2016040896
https://patents.google.com/patent/WO2016040896A1/en
References
- ^ Jump up to:a b c d e http://adisinsight.springer.com/drugs/800035241
- ^ http://www.pharmaceutical-technology.com/news/newsfda-grants-fast-track-status-innocrins-seviteronel-treat-metastatic-crpc-4770025
- ^ Yin L, Hu Q, Hartmann RW (2013). “Recent progress in pharmaceutical therapies for castration-resistant prostate cancer”. Int J Mol Sci. 14 (7): 13958–78. doi:10.3390/ijms140713958. PMC 3742227. PMID 23880851.
- ^ Jump up to:a b Stein MN, Patel N, Bershadskiy A, Sokoloff A, Singer EA (2014). “Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer”. Asian J. Androl. 16 (3): 387–400. doi:10.4103/1008-682X.129133. PMC 4023364. PMID 24759590.
- ^ Jump up to:a b Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ (2014). “Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors”. Bioorg. Med. Chem. Lett. 24 (11): 2444–7. doi:10.1016/j.bmcl.2014.04.024. PMID 24775307.
- ^ Jump up to:a b c d Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME (2015). “Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer”. Mol. Cancer Ther. 14 (1): 59–69. doi:10.1158/1535-7163.MCT-14-0521. PMID 25351916.
- ^ Jump up to:a b c Stephen Neidle (30 September 2013). Cancer Drug Design and Discovery. Academic Press. pp. 341–342. ISBN 978-0-12-397228-6.
- ^ Jump up to:a b Wm Kevin Kelly; Edouard J. Trabulsi, MD; Nicholas G. Zaorsky, MD (17 December 2014). Prostate Cancer: A Multidisciplinary Approach to Diagnosis and Management. Demos Medical Publishing. pp. 342–. ISBN 978-1-936287-59-8.
- ^ http://www.who.int/medicines/publications/druginformation/innlists/RL76.pdf
Further reading
- Gomez L, Kovac JR, Lamb DJ (2015). “CYP17A1 inhibitors in castration-resistant prostate cancer”. Steroids. 95: 80–7. doi:10.1016/j.steroids.2014.12.021. PMC 4323677. PMID 25560485.
- Bambury RM, Rathkopf DE (2015). “Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide”. Urol. Oncol. 34: 348–55. doi:10.1016/j.urolonc.2015.05.025. PMID 26162486.
External links[
|
|
| Clinical data | |
|---|---|
| Synonyms | VT-464; INO-464 |
| Routes of administration |
By mouth |
| Drug class | Androgen biosynthesis inhibitor; Nonsteroidal antiandrogen |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C18H17F4N3O3 |
| Molar mass | 399.339 g/mol g·mol−1 |
| 3D model (JSmol) | |
References
-
Innocrin Pharmaceuticals Created as a Spin-out of the Prostate Cancer Program from Viamet Pharmaceuticals.
-
Viamet Pharmaceuticals and the Novartis Option Fund Enter Agreement for Development of Novel Metalloenzyme Inhibitors.
-
Innocrin Pharmaceuticals, Inc. Granted SME Status Designation by the European Medicines Agency.
-
A Single arm, open label, signal seeking, Phase II a trial of the activity of seviteronel in patients with androgen receptor (AR) positive solid tumours
-
Innocrin Pharmaceuticals and the Prostate Cancer Foundation (PCF) Join Forces for Innovative Phase 2 Clinical Study.
-
A Phase 2 Open-label Study to Evaluate the Efficacy and Safety of Seviteronel in Subjects With Castration-Resistant Prostate Cancer Progressing on Enzalutamide or Abiraterone
-
Innocrin Pharmaceuticals, Inc. Granted Fast Track Designation by FDA for VT-464 Treatment of Patients with Metastatic Castrate-resistant Prostate Cancer.
-
Innocrin Pharmaceuticals, Inc. Begins Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer and Expands Two Phase 2 Studies of Seviteronel in Men with Metastatic Castrate-resistant Prostate Cancer.
-
A Phase 2 Open-Label Study to Evaluate the Efficacy and Safety of VT-464 in Patients With Metastatic Castration Resistant Prostate Cancer Who Have Previously Been Treated With Enzalutamide, Androgen Receptor Positive Triple-Negative Breast Cancer Patients, and Men With ER Positive Breast Cancer
-
Innocrin Pharmaceuticals Inc. to Present Interim Results from Its Phase 1/2 Prostate Cancer Clinical Study and Preclinical Results That Demonstrate VT-464 Efficacy in a Clinically-Relevant Enzalutamide-Resistant Mouse Model.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of Seviteronel in Subjects With Castration-Resistant Prostate Cancer
-
A Phase 1/2 Open-Label, Multiple-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Once-Daily VT-464 in Patients With Castration-Resistant Prostate Cancer
-
Viamet Pharmaceuticals Appoints Former Novartis Executive Marc Rudoltz, M.D. as Chief Medical Officer.
-
VIAMET PHARMACEUTICALS AND THE NATIONAL INSTITUTES OF HEALTH TO JOINTLY DEVELOP NOVEL VIAMET COMPOUND.
-
Viamet Pharmaceuticals Initiates Phase 1/2 Clinical Trial of Novel Prostate Cancer Therapy, VT-464.
-
Viamet Pharmaceuticals to Present at the 32nd Annual J.P. Morgan Healthcare Conference.
-
VIAMET PHARMACEUTICALS TO PRESENT AT THE 31st Annual J.P. MORGAN HEALTHCARE CONFERENCE.
-
Innocrin Pharmaceuticals, Inc. Initiates Phase 2 Castration-Resistant Prostate Cancer (CRPC) Study in Men Who Have Failed Enzalutmaide or Abiraterone.
-
Innocrin Pharmaceuticals Appoints Fred Eshelman, PharmD as CEO and is Granted Fast Track Designation by FDA for Seviteronel Treatment of Women with Triple-negative Breast Cancer and Women or Men with Estrogen Receptor-positive Breast Cancer.
-
Gucalp A, Bardia A, Gabrail N, DaCosta N, Danso M, Elias AD, et al. Phase 1/2 study of oral seviteronel (VT-464), a dual CYP17-lyase inhibitor and androgen receptor (AR) antagonist, in patients with advanced AR positive triple negative (TNBC) or estrogen receptor (ER) positive breast cancer (BC). SABCS-2016 2016; abstr. P2-08-04.
Available from: URL:http://www.abstracts2view.com/sabcs/view.php?nu=SABCS16L_1479
-
Innocrin Pharmaceuticals Presents Data from the Ongoing Phase 2 Trial of Seviteronel in Estrogen Receptor-positive or Triple-negative Breast Cancer (CLARITY-01) at the San Antonio Breast Cancer Symposium.
-
Innocrin Pharmaceuticals, Inc. Appoints Edwina Baskin-Bey, MD as Chief Medical Officer and Expands the Ongoing Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer (TNBC).
-
Innocrin Pharmaceuticals, Inc. Raises $28 Million in Series D Financing.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Efficacy of Seviteronel in Subjects With Advanced Breast Cancer
-
Speers CW, Chandler B, Zhao S, Liu M, Wilder-Romans K, Olsen E, et al. Radiosensitization of androgen receptor (AR)-positive triple-negative breast cancer (TNBC) cells using seviteronel (SEVI), a selective CYP17 lyase and AR inhibitor. ASCO-2017 2017; abstr. e12102.
Available from: URL: http://abstracts.asco.org/199/AbstView_199_193240.html
-
Innocrin Pharmaceuticals, Inc. Appoints Charles F. Osborne Jr. as its Chief Financial Officer.
-
Viamet Pharmaceuticals Secures $18 Million Financing.
-
Viamet Pharmaceuticals Raises $4 Million Round of Financing.
///////////SEVITERONEL, VT-464, INO-464, VT 464, INO 464, Phase II, Breast cancer, Prostate cancer, Solid tumours, viamet, CANCER, севитеронел , سيفيتيرونيل , 赛维罗奈 ,
C1(=CN=NN1)C(C1=CC2=C(C=C1)C=C(C(=C2)OC(F)F)OC(F)F)(C(C)C)O