
BI-882370
XP-102
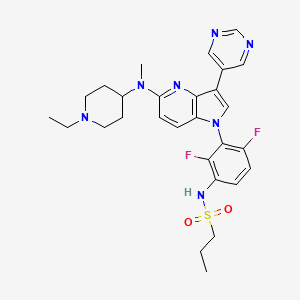
N-(3-(5-((1-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-2,4-difluorophenyl)propane-1-sulfonamide
CAS 1392429-79-6
Chemical Formula: C28H33F2N7O2S
Molecular Weight: 569.68
Elemental Analysis: C, 59.03; H, 5.84; F, 6.67; N, 17.21; O, 5.62; S, 5.63
BI 882370 is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the BRAF kinase. BI 882370 inhibits proliferation of human BRAF-mutant melanoma cells with 100× higher potency (1-10 nmol/L) than vemurafenib.
Xynomic, under license from Boehringer Ingelheim , is investigating for treating BRAF mutant cancers, including colorectal cancer and melanoma; in October 2017, preclinical data were reported in the melanoma and colorectal cancer settings.
- Originator Boehringer Ingelheim
- Developer Boehringer Ingelheim; Xynomic Pharmaceuticals
- Class Antineoplastics; Piperidines; Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Proto oncogene protein b raf inhibitors
- Preclinical Colorectal cancer; Malignant melanoma
- 20 Dec 2018 Xynomic Pharma plans a phase Ib trial for Colorectal cancer (in combination with BI 860585) in third quarter of 2019
- 01 Jun 2018 Xynomic Pharmaceuticals plans a phase I trial for Colorectal cancer and Malignant melanoma in 2018 or 2019
- 06 Nov 2017 Chemical structure information added
-
US8889684
PATENT
WO2012104388

PATENT
Novel crystalline salts (monosuccinate salt), designated as Form A, of BI-882370 and their substantially anhydrous and non-solvated, processes for their preparation and compositions comprising them. Also claimed are their use as a RAF kinase Inhibitor, for the treatment of cancers and other diseases, such as infections, inflammations and autoimmune diseases.
The compound N-(3-(5-((l -ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3, 2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide (BI 882370), having Formula I:
I
is a RAF kinase inhibitor useful in the treatment of various diseases including cancer. The compound of Formula I, as well as its preparation and use, have been described in
WO/2012/104388, which is incorporated herein by reference in its entirety.
The RAS-RAF-MAPK (mitogen-activated protein kinase) signaling pathway plays a critical role in transmitting proliferation signals generated by the cell surface receptors and cytoplasmic signaling elements to the nucleus. Constitutive activation of this pathway is involved in malignant transformation by several oncogenes. Activating mutations in RAS
occur in approximately 15 % of cancers, and recent data has shown that B-RAF is mutated in about 7% of cancers (Wellbrock et al, “The RAF proteins take centre stage”, Nature Rev. Mol. Cell Biol., 2004, 5, 875-885), identifying it as another important oncogene in this pathway. In mammals, the RAF family of serine/threonine kinases comprises three members: A-RAF, B-RAF and C-RAF. However, activating mutations have so far been only identified in B-RAF underlining the importance of this isoform. It is believed that B-RAF is the main isoform that couples RAS to MEK, and that C-RAF and A-RAF signal to ERK only to fine-tune cellular responses (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). The most common cancer mutation in B-RAF results in a valine to glutamic acid exchange at position 600 of the protein (V600E), which dramatically enhances B-RAF activity, presumably because its negative charge mimics activation loop phosphorylation (Wan et al , “Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867). The highest incidence of B-RAF V600 mutations occurs in malignant melanoma (39%), thyroid cancer (46%), colorectal cancer (10%), biliary tract cancer (10%), prostate cancer (4%), ovary cancer (3%) and non-small cell lung cancer (2%), but they also occur at a low frequency in a wide variety of other cancers (frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v.53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Literature supported the hypothesis that B-RA 600E mutated tumor cells seem to rely heavily on the continued activation of this pathway – a phenomenon termed “oncogene addiction” – whereas normal B-RAFwt cells use a broader range of signals. This provides an Achilles’ heel that can be exploited
therapeutically by treating patients with somatically mutated B-RAFV600E using orally available B-RAF inhibitors.
The key role of B-RAF V600E in aberrant ERK signaling and consequently oncogenesis has been demonstrated in several independent experimental approaches such as
overexpression of oncogenic/mutated B-RAF in vitro and in vivo (Wan et al., Cell, 2004, 116, 855-867; Wellbrock et al, Cancer Res. 2004, 64: 2338-2342), siRNA knock-down in vitro (Karasarides et al., Oncogene, “V599EB-RAF is an oncogene in melanocytes”, 2004, 23, 6292-6298) or in inducible short-hairpin RNA xenograft models where gain-of-function B-RAF signaling was found to be strongly associated with in vivo tumorigenicity (Hoeflich et al, “Oncogenic BRAF is required for tumor growth and maintenance in melanoma models”, Cancer Res., 2006, 66, 999-1006).
Treatment of B-RAFV600E mutated melanoma or colon carcinoma cells induces a B-RAF inhibition phenotype (e.g. reduction of phospho-MEK and phospho-ERK levels, reduction of cyclin D expression and induction of p27 expression). Consequently, these cells are locked in the Gl -phase of the cell cycle and do not proliferate.
Clinical proof of mechanism and proof of concept has been established for treating in cancer in B-RAFV600E mutated melanoma patients treated with Zelboraf®, B-RAF inhibitor (PLX-4032, vemurafenib, from Plexxikon/Daiichi Sankyo/Roche. Bollag et al., “Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma”, Nature, 2010, 467(7315), 596-9.; Flaherty et al, New Engl. J. Med., “Inhibition of Mutated, Activated BRAF in Metastatic Melanoma”, 2010, 363, 809-819; Chapman et al. “Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation”, New Engl. J. Med, 2011, 364:2507-2516. Favorable response rates were observed in both Phase I and Phase III clinical trials. It was reported, that melanoma patients carrying a B-RAFV600K mutation also do respond to therapy (Rubinstein et al, “Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032”, J. Transl. Med , 2010, 8, 67).
The most frequent B-RAF mutation is the exchange at amino acid position 600 from valine to glutamate with more than 90% frequency of all B-RAF mutations (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885), the second most frequent mutation is an alteration from valine to lysine, other mutations were found with lower frequency at that position (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885 and frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Additional mutations were found at e.g. the glycine rich loop (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). Not all of these rather rare mutations seem to lead to direct activation of B-RAF (Wan et al. ,
“Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867).
The compound of Formula I is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the B-RAF kinase. The compound inhibited proliferation of human B-RAF-mutant melanoma cells with 100 times higher potency (1-10 nmol/L) than vemurafenib, whereas wild-type cells were not affected at 1,000 nmol/L. A solution of the compound administered orally was efficacious in mouse models of B-RAF-mutant melanomas and colorectal carcinomas, and at 25 mg/kg twice daily showed superior efficacy compared with vemurafenib, dabrafenib, or trametinib. The compound was also active in A375 melanoma-bearing mice that were resistant to vemurafenib, particularly when dosed in combination with trametinib. Mice treated with the compound did not show any body weight loss or clinical signs of intolerability, and no pathologic changes were observed in several major organs investigated, including skin. Furthermore, in a pilot study in rats (up to 60 mg/kg daily for 2 weeks), the compound lacked toxicity in terms of clinical chemistry, hematology, pathology, and toxicogenomics. These results are described in Waizenegger et al., Mol. Cancer Ther., 2016, 75(3); 354-65, which is incorporated herein by reference in its entirety.
For the manufacture, purification, and formulation of a drug, it may be advantageous to employ a form of the drug having superior stability or other desirable formulation property exhibited by, for example, one or more salt or crystalline forms of the drug. Formation of salts of basic or acidic drugs can sometimes provide forms of the drug that have
advantageous properties such as solubility, non-hygroscopicity, crystallinity, and other physical properties that advantageous for formulating the drug. On the other hand, discovering a suitable salt or other crystalline form that is suitable for formulation is difficult, since there are numerous variables in the formation of a salt or crystalline form. These include the existence of numerous possible acids and bases that might be used as a counter-ion, various stoichiometric ratios that may be possible for combining a given basic or acid drug with an acid or base counter-ion, a wide variety of solvents and solvent systems
(including combinations of solvents) that potentially can be used to attempt to form salts or crystalline forms, and a variety of conditions (such as temperature or heating or cooling conditions) under which salts or crystalline forms may be generated. All of these variables of which may affect the properties of the salts or crystalline forms that might be obtained. Salts or solid forms may also have a variety of properties that render them unsuitable for drug development and formulation such as lack of crystallinity (amorphous forms), the presence or formation of multiple crystalline forms, which may interconvert and/or have different properties (polymorphism), lack of aqueous solubility, hygroscopicity, or stickiness of the solid. Furthermore, the formation of salts and crystalline forms and their properties are generally very unpredictable.
Accordingly, the crystalline salt forms of the compound of Formula I provided herein help satisfy the ongoing need for the development of a RAF kinase inhibitor for the treatment of serious diseases.
Preparation of A^-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l- amide (BI 882370)
Step 1. 4-(6-Methyl-5-nitro-pyridin-2-yl)-piperazine-l-carboxylic acid tert-butyi ester
(3)
1 2 3
DIPEA (62.82 mL, 0.435 mol) is added to the solution of 6-chloro-3-nitro-2-methylpyridine (1) (50 g, 290 mmol) and N-Boc-piperazine (2) (53.95 g, 290 mmol) in dry MeCN (200 mL) and stirred for 4 h at 50 °C. After the reaction is finished the reaction mixture is diluted with MeCN and water and stirred for 30 min. The precipitated product is collected by filtration, washed with water and the solid is dried in vacuo.
Step 2. 4- [6-((£’)-2-Dimethylamino-vinyl)-5-nitro-pyridin-2-yl] -piperazine- 1-carboxylic acid
To a stirred solution of 4-(6-methyl-5-nitro-pyridin-2-yl)-piperazine- 1-carboxylic acid tert-butyl ester (3) (13 g, 40.3 mmol) in DMF (35 mL) is added N,N-dimethylformamide dimethylacetal (14.47 g, 121 mmol) and stirred in argon atmosphere for 36 h at 90 °C.
Additional 1.5 eq. of N^V-dimethylformamide dimethylacetal is added and stirred for 12 h at 90 °C. The reaction mixture is poured into water and extracted with DCM. The combined organic layers are washed with water, dried over anhydrous Na2S04 and concentrated in vacuo. The residue is used without further purification for the next step.
Step -(lH-pyrrolo[3,2-Z>]pyridin-5-yl)piperazine-l-carboxylic acid tert-butyl ester (5)
4 5
4-[6-((i?)-2-Dimethylairdno-vinyl)-5-nitro-pyridin-2-yl]-piperazine-l-carboxylic acid tert-butyl ester (36.4 g, 96 mmol) is taken up in MeOH, Pd/C (0.56 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 60 psi for 16 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by column chromatography viaNP MPLC. The product containing fractions of compound (5) (HPLC-MS method B: tRet. = 1.55 min.; MS (M+H)+ = 303) are combined and evaporated in vacuo.
Step 4. N- -Amino-2,6-difluorophenyl)acetamide (7)
6 7
Compound (6) (55.0 g, 254 mmol) is taken-up in MeOH (1.0 L). Pd/C (10.0 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 200 psi for 3 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by NP-MPLC on silica gel using DCM/MeOH (96:4) as eluent. The product containing fractions of the aniline intermediate (HPLC-MS method B: tRet. = 0.25 min.; MS (M-H)“ = 185) are combined and evaporated.
Step 5. N- -Difluoro-3-(propylsulfonamido)phenyl)acetamide (9)
To the aniline intermediate (35.0 g, 188 mmol) in DCM (100 mL) pyridine (6.6 mL, 75 mmol) and ^-propane sulfonyl chloride (8) (29.5 mL, 263 mmol) are added and the mixture is stirred at rt for 16 h. The reaction mixture is diluted with EtOAc (200 mL), washed with H2O and HC1 (aq., 1 N) and the layers are separated, dried over MgS04 and evaporated to yield the sulfonamide (9) which was used without further purification.
Step 6. N-
9 10
The sulfonylated aniline (9) (38.0 g, 130 mmol) is taken-up in EtOH (250 mL), H2O (200 mL) and concentrated hydrochloric acid (200 mL) and heated to 80 °C for 2 h. The reaction mixture is concentrated under reduced pressure, aqueous NaOH (4 N) is added until pH = 6 is reached and the mixture is extracted 2 x with DCM. The combined organic layer is washed with brine, dried over MgS04, filtered and evaporated to yield the deacylated aniline (10) (HPLC-MS method B: tRet. = 0.22 min.; MS (M-H)“ = 249) as a hydrochloride which was used without further purification.
Step 7. N-(2 -Difluoro-3-iodophenyl)propane-l-sulfonamide (11)
10 11
The hydrochloride of compound (10) is taken-up in DCM and extracted with NaHCCb solution. The organic layer is dried over MgSCn, filtered and evaporated. To the free base (10) (3.55 g, 14.21 mmol) in TFA (80 mL) at 0 °C is added NaNC (1.96 g, 28.4 mmol) in small portions and the mixture is stirred for 30 min. KI (23.83 g, 142 mmol) is added and stirring is continued for additional 15 min. The reaction mixture is diluted with Et^O and stirred for 1 h. Na2S203 solution (semiconc.) is added and the mixture is extracted 3 x with Et20. The combined organic layer is dried over MgSCn, filtered and concentrated in vacuo. The residue is purified by column chromatography via NP-MPLC. The product containing fractions of compound (11) (HPLC-MS method A: tRet. = 1.58 min.; MS (M-H)“ = 360) are combined and evaporated in vacuo.
Step 8. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-lH-pyrrolo [3,2-b] pyridin-5-yl)
12
The lH-pyrrolo [3,2-*] pyridine (5) (10.0 g, 30.27 mmol), sulfonamide (11) (16.4 g,
45.4 mmol), Cul (576 mg, 3.03 mmol), ^^-(l ^^^-^N’-bismethyl-l^-cyclohexandiamine
(1.91 mL, 12.1 mmol) and CS2CO3 (29.6 g, 90.85 mmol) are taken-up in dry toluene (3 mL) and the resulting mixture is flushed with argon and stirred for 16 h at 120 °C. After the addition of further Cul (576 mg, 3.03 mmol), trans-(\R,2R)-N,N’-bismet y 1-1,2-cyclohexandiamine (1.91 mL, 12.1 mmol) and CS2CO3 (20.0 g, 60.0 mmol) the reaction mixture is stirred for further 24 h. The solvent is removed in vacuo, the residue is taken up in DCM and extracted with NaHCC solution (semiconc). The organic layer is dried over MgS04, filtered, the solvent is removed in vacuo and the residue is purified viaNP-MPLC. The product containing fractions of (12) (HPLC-MS method C: teet. = 1.62 mia; MS (M+H)+ = 564) are combined and the solvent is removed in vacuo.
Step 9. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-iodo-lH-pyrrolo[3,2-b]pyridin-5 3)
To a solution of sulfonamide (12) (1.078 g, 1.9 mmol) in DMF (4 mL)/THF (100 μί) is added NIS (474 mg, 2.1 mmol) and the mixture is stirred for 1 h at rt. The reaction mixture is diluted with 30 mL DCM and extracted with NaHCCb solution (semiconc). The combined organic layer is dried over MgSCn, filtered and concentrated under reduced pressure. The residue is purified by column chromatography via RP HPLC. The product containing fractions of (13) (HPLC-MS method B: tRet. = 2.035 mia; MS (M+H)+ = 688) are freeze dried.
Step 10. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-b]pyridin-5-yl)(methyl)amino)piperidine-l-carboxylic acid tert-butyi ester (15)
13 15
Sulfonamide (13) (770 mg, 1.12 mmol), pyrimidin-5-yl-boronic acid (14) (194 mg, 1.57 mmol), Pd(dppf)Cl2 (82 mg, 0.11 mmol), LiCl (142 mg, 3.35 mmol) and Na2C03 (294 mg, 2.8 mmol) are taken-up in dioxane/LhO (2: 1 mixture, 12 mL), and the resulting mixture is flushed with argon and stirred for 1 h at 100 °C. The reaction mixture is diluted with DCM and extracted with NaHCCb solution (semi-concentrated). The organic layer is dried over MgS04, filtered, Isolute® is added, the solvent is removed in vacuo and the residue is purified via RP HPLC. The product containing fractions of (15) (HPLC-MS method C: tRet. = 2.149 min.; MS (M+H)+ = 642) are freeze dried.
Step 11. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)- 1H-pyrrolo[3,2-b]pyridin-l-yl)phenyl)propane-l-sulfonamide
15 16
To a solution of example compound (15) (154 mg, 0.24 mmol) in DCM/MeOH (1 : 1, 4 mL) is added HC1 (in dioxane, 4 N, 2 mL) and the mixture is stirred for 3 h at rt. The solvent is removed in vacuo. Obtained compound (16) (HPLC-MS method B: tRet. = 1.02 min.; MS (M+H)+ = 542) is used without further purification.
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-diflu
Compound I was obtained from compound (16) by reductive alkylation with acetaldehyde (40% in iPrOH) in the presence of 1.5 eq. sodium acetoxyborohydride in iPrOH. The crude product was recrystallized from ethanol to obtain the title compound in 84% yield.
Scale-Up Synthesis of A/-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (BI 882370)
Step 1. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[
15 16
Isopropanol (8.83 kg) and compound (15) (1.80 kg, 2.8 mol) were added into a reactor, and the mixture was stirred and heated to 55-60 °C. Concentrated hydrochloric acid (2.76 kg, 28 mol) was dropped into the reactor over than 20 min. at 60-65 °C. Then, the reaction mass was heated to 60-70 °C and held for 1 h. The conversion was monitored by HPLC, and reached about 99.5% after about 1 h.
The reaction mass was cooled and the isopropanol was removed by distillation under reduced pressure at not more than 50 °C. A brown oil was obtained, dissolved into water (6.75 kg) and washed by extraction with ethyl acetate (2.02 kg) at 20-30 °C. The water-phase was cooled to 15-20 °C. The pH was adjusted to 8.0-8.5 with 10% aqueous NaOH solution (-8.0 kg) at 20-30°C. The mixture was stirred for 3-4h at 20-30°C with the pH adjusted to 8.0-8.5 by addition of 10% NaOH solution every half-hour. The product was isolated by filtration and the cake washed with water (3.6 kg). The solid was dried under vacuum at 45-50 until the water content was not more than 5.5%. This provided about 1.64 kg of crude compound (16) (yield 108% of theoretical; the crude product containing water and NaCl detected). The crude product was used directly).
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrr -Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (I)
Bl 878426 Bl 882370
Process:
Dichloromethane (19.88 kg) and compound (16) (1.5kg, 2.77mol) were added into a reactor, and the mixture was stirred and cooled to 0-10°C under a nitrogen atmosphere. Sodium triacetoxyborohydride (95%, 0.93 kg, 4.16 mol) was added into the mixture at 0-10°C. The mixture was stirred for 20-30 min. at 0- 10°C. Acetaldehyde in DCM (40%,
1.07 kg, 9.71 mol) added into the mixture slowly over 2 h at 0-10 °C. The reaction mixture was stirred at 0-10 °C under a nitrogen atmosphere for 0.5-lh. The conversion was monitored by HPLC, and reached about 99.5% after about 0.5-1 h.
Water (15 kg) was added into the reaction mass at a temperature below 15 °C. The mixture was stirred at 15-30 °C for 20-30 min. Aqueous ammonia (25%, 1.13 kg, 16.61 mol) was added into the mixture and the mixture was then stirred for 0.5 h. The organic phase was separated and then washed by extraction with water (15 kg) at 20-25 °C. Activated charcoal (0.15 kg) was added into the organic phase. The mixture was stirred for 1 h and then filtered. The filtrate was concentrated under reduced pressure at not more than 40°C, and compound (I) (1.58 kg, 100% yield) was obtained as a foamy solid.
Investigation of the Crystallinity of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide Free Base
Investigation of the crystallinity of N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(py rimidin-5-y 1)- lH-pyrrolo[3 ,2-b] pyridin- 1 -y l)-2,4-difluoropheny l)propane- 1 -sulfonamide free base, obtained by recrystallization from aqueous ethanol, which was used as a starting material to investigate salt formation showed that the compound had low crystallinity, as seen in FIG. 1.
Investigation of Salt forms of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide
The compound N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3 ,2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide was combined with various acids in various solvent systems.
A 96-well master plate was charged by dosing compound in MeOH (stock solution) with a concentration of approx. 40 mg/mL. This plate was placed in a vacuum oven for liquid removal to obtain the same amount of solid material in each well. Subsequently different solvents/solvent mixtures and the acids were added to the solid material in each well (approx. 500μί) and the whole plate was heated up to 50 °C for 2 hours while stirring (using a small stirring bar added to each well).
The acids used were as shown in Table 1. The solvents used were as shown in Table 2. Crystallinity of salts obtained either by the slurry experiment or crystallization by evaporation.
To investigate crystal formation by a slurry experiment, the plate was allowed to cool and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 2A and images of XRPD performed on the salt from each of the master plate wells, showing the crystallinity of the salts formed, is shown in FIG. 2B.
To investigate crystal formation by an evaporation experiment, after the heating period, the solutions were filtered at the same temperature (50 °C) using a preheated filter plate to ensure that no non-dissolved material can be transferred into the other crystallization plates. The filtrate was dispensed into an evaporation plate (approx.. 200μί). The solvents were allowed to evaporate, and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 3A and images of XRPD performed on the salt from each of the evaporation plate wells, showing the crystallinity of the salts formed, is shown in FIG. 3B.
Table 1. Salts Used for Salt Form Investigation
Table 2. Solvents Used for Salt Form Investigation
REFERENCES
1: Waizenegger IC, Baum A, Steurer S, Stadtmüller H, Bader G, Schaaf O, Garin-Chesa P, Schlattl A, Schweifer N, Haslinger C, Colbatzky F, Mousa S, Kalkuhl A, Kraut N, Adolf GR. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol Cancer Ther. 2016 Mar;15(3):354-65. doi: 10.1158/1535-7163.MCT-15-0617. Epub 2016 Feb 25. PubMed PMID: 26916115.
/////////////// BI-882370, BI 882370, BI882370, XP-102, Boehringer Ingelheim, Xynomic Pharmaceuticals, Preclinical, Colorectal cancer, Malignant melanoma
CCN1CCC(CC1)N(C)c3ccc4n(cc(c2cncnc2)c4n3)c5c(F)ccc(NS(=O)(=O)CCC)c5F