Solriamfetol hydrochloride
FDA APPROVED 2019/3/20, Sunosi
ソルリアムフェトル塩酸塩; R228060, R 228060
| Formula |
C10H14N2O2. HCl
|
|---|---|
| CAS |
178429-65-7 HCL
|
| Mol weight |
230.6913
|
- Developer Jazz Pharmaceuticals plc; SK biopharmaceuticals
- Class Carbamates; Sleep disorder therapies; Small molecules
- Mechanism of Action Adrenergic uptake inhibitors; Dopamine uptake inhibitors
- Orphan Drug Status Yes – Narcolepsy
- Registered Hypersomnia
- Discontinued Depressive disorders
- 26 Mar 2019 Discontinued – Phase-I for Depressive disorders (Adjunctive treatment) in USA (PO) (Jazz Pharmaceuticals pipeline, March 2019)
- 20 Mar 2019 Registered for Hypersomnia (excessive daytime sleepiness) in patients with obstructive sleep apnoea and narcolepsy in USA (PO) – First global approval
- 20 Mar 2019 US FDA approves solriamfetol to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea(OSA)
- New Drug Application (NDA): 211230
Company: JAZZ PHARMA IRELAND LTD
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1]
Common side effects include headache, nausea, anxiety, and trouble sleeping.[1] It is a norepinephrine–dopamine reuptake inhibitor(NDRI). It is derived from phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[2]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]Aerial ran two Phase II trials of the drug in narcolepsy[4] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticalspaid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double digit royalties to SK.[3][5]
In March 2019 the FDA accepted SK’s and Jazz’ NDA for use of solriamfetol to treat excessive sleepiness in people with narcolepsy or obstructuve sleep apnea; the drug has an orphan designation for narcolepsy.[3][6]
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[6]
Research
Solriamfetol had also been tested in animal models of depression, but as of 2017 that work had not been advanced to clinical trials.[7]
PATENT
WO 9607637
https://patents.google.com/patent/WO1996007637A1/e
Organic alkyl carbamates have been effectively used for controlling various central nervous system (CNS) disorders. For example, U.S. Pat. Nos . 2,884,444, 2,937,119 and 3,313,697 disclose function of carbamate in CNS disorders, especially as antiepileptic and centrally acting muscle relaxant.
Phenylethylamine derivatives, one important class of therapeutical medicines useful for managing CNS diseases, have been used mainly to treat obesity, narcolepsy, minimal brain dysfunction and mild depression.
Recent design of pharmacologically useful compounds has been based on amino acids or the derivatives thereof, which is mainly attributable to the fact that many of the compounds found in biological systems come from amino acids or the derivatives thereof. In addition, in most cases, the function of a pharmaceutically useful compound is effected after it binds to an enzyme or receptor, which may trigger the regulatory mechanisms of the enzyme or receptor.
REACTION SCHEME I
REACTION SCHEME II
REACTION SCHEME III
EXAMPLE I
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
In a 500 mL RB flask equipped with a mechanical stirrer and a dropping funnel, D-phenylalaninol (45.4 g, 300 mmol) was dissolved in 220 mL of distilled water, and cooled in an ice-bath. The pH of the solution was adjusted with 50 % sodium hydroxide to 14. Benzyl chloroformate (49.3 mL, 345 mmol) was charged into the dropping funnel and added slowly to the well stirred solution over 0.5 hr. After the completion of the addition, the reaction mixture was stirred for 1 hr. at 0 *C. The product precipitated from the reaction mixture as a white solid. It was collected by filtration and washed completely with distilled water. After being dried in vacuo, the solid thus obtained weighed 104 grams without any further purification: 99.8% Yield.
Melting point = 90 – 92 *C
[α]D20 = + 43.4 (c = 1.0, EtOH)
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.35; H, 6.71; N,4.91
EXAMPLE II
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
carbamate
In a 500 mL RB flask, N-benzyloxycarbonyl-D- phenylalaninol (13.56 g, 50 mmol) was charged with antipyrine (11.29 g, 60 mmol) in 250 mL of dry THF under a nitrogen atmosphere. The reaction mixture was cooled in an ice-bath and phosgene (30.3 mL of 1.93 M solution in toluene, 58.5 mmol) was added quickly while vigorously stirring. After stirring for 1 hr. , the formation of a corresponding chloroformate from the starting material was monitored by TLC. The chloroformate solution thus prepared, was slowly added to a well stirred and ice-chilled aqueous ammonium hydroxide solution (75 mL, 28-30 %, 1,190 mmol) via cannula over 0.5 hr. The resulting reaction mixture was stirred for an extra 0.5 hr. The organic phase separated was collected. The aqueous phase was extracted twice with methylene chloride (100 mL). The combined organic phase was washed with brine (50 mL), dried over sodium sulfate, and concentrated to yield 17.8 g (113%) of foamy solid. It was purified a flash column chromatography to give 14.8 g of the title compound, white solid: 94% Yield.
Melting point = 121 – 125 *C
[α]D20 = + 28.6 (c = 2.0, EtOH)
Analysis calc. : C, 65.84; H, 6.14; N, 8.53
Found: C, 66.68; H, 6.21; N, 7.80
EXAMPLE III
Preparation of D-Phenylalaninol carbamate hydrochloric
acid salt In a 160 mL Parr reactor, N-benzyloxycarbonyl-D-phenylalaninol carbamate (9.43 g) was added with 75 mL of anhydrous methanol and 10 % palladium on charcoal (0.32 g). Then, the reactor was closed and purged with hydrogen for 1 in. The reaction was completed in 2 hrs . under 40 psi pressure of hydrogen at 45 #C. The catalyst was filtered off. Thereafter, the organic layer was concentrated into 5.97 g (102 %) of pale yellow thick liquid. The liquid was poured in 50 mL of anhydrous THF and cooled to 0 “C. Anhydrous hydrogen chloride gas was then purged through the solution with slowly stirring for
0.5 hr. 50 mL of anhydrous ether was added, to give a precipitate. Filtration with THF-ether (1:1) mixture provided 6.1 g of the title compound as a white solid: 88 % Yield.
Melting point = 172 – 174 “C
[α]D20 = – 12.9 (c = 2.0, H20)
Analysis calc. : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.90; H, 6.60; N, 12.15; Cl ,
15.52
EXAMPLE IV
Preparation of N-benzyloxγcarbonyl-L-Phenγlalaninol
The title compound was prepared in the same manner as that of Example I, except that (L)-phenylalaninol was used as the starting material.
Melting point = 90 – 92 *C
[α]D20 = – 42.0 (c = 1.0, EtOH)
Analysis calc. : C, 71.56; H, 6.71; N,4.91
Found: C, 70.98; H, 6.67; N,4.95
EXAMPLE V
Preparation of -N-benzyloxycarbonyl-L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-L-phenylalaninol was used as the starting material.
Melting point = 121 – 128 ‘C
[α]D20 = – 28.9 (c = 2.0, EtOH)
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.45; H, 6.15; N, 8.32
EXAMPLE VI
Preparation of L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-L-phenylalaninol carbamate was used as the starting material.
Melting point = 175 – 177 *C [α]D20 = + 13.1 (c = 1.0, H20)
Analysis calc : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.95; H, 6.58; N, 12.09; Cl , 15.37
EXAMPLE VII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
The title compound was prepared in the same manner as that of Example I, except that (D,L)-phenylalaninol was used as the starting material.
Melting point = 72 – 75 #C
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.37; H, 6.74; N,4.84
EXAMPLE VIII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-D,L-phenylalaninol was used as the starting material.
Melting point = 130 – 133 *C
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.85; H, 6.14; N, 8.49 EXAMPLE IX
Preparation of D,L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-D,L-phenylalaninol carbamate was used as the starting material.
Melting point = 163 – 165 *C
Analysis calc: C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.92; H, 6.56; N, 11.95; Cl , 15.82
PATENT
US 20050080268
PATENT
WO 2018133703
https://patents.google.com/patent/WO2018133703A1/en
Excessive daytime sleepiness (Excessive Daytime Sleepiness, EDS) or pathological somnolence refers to excessive daytime sleep and wakefulness associated with various sleep disorders. These disorders can be the basis for a sleep disorder or sleep have side effects caused by some other medical conditions. Excessive daytime sleep, also known as narcolepsy, sleep clinics is seen mainly in patients with disease that affects 12% of the general population. EDS patients may be manifested as mental distress, poor work or school performance, increasing the risk of accidents, the impact of EDS can debilitating, even life-threatening.
R228060, also known JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor, originally developed by R & D, SK biopharmaceutical, 2014 Sir ownership of the pharmaceutical compound. R228060 has the potential to treat narcolepsy and sleep apnea syndrome, in three multi-center study in two global reached the primary endpoint, and achieved positive results, significantly improved adult obstructive sleep apnea patients excessive sleepiness in patients with narcolepsy and excessive sleep problems.
R228060 chemical name is O- carbamoyl – (D) – phenylalaninol, as shown in the structural formula of formula (I):

Solid Form different chemicals, can cause varying their solubility and stability, and thus affects the absorption and bioavailability of the drug, and can lead to differences in clinical efficacy. Improve the candidate compound has a solubility by salt way become an important means of drug development. Compared to the free form of the drug, suitable pharmaceutically acceptable salts can improve the solubility of the drug type, increased physical and chemical stability, and also to improve the drug-salt having a melting point, hygroscopicity, crystal type and other physical properties, further development of the pharmaceutical dosage form It plays an important role. Patent Document WO1996007637A1 discloses R228060 hydrochloride and its preparation method, and other characteristics of the obtained having a melting point of 172-174 deg.] C as a white solid, the solid was not given in the text data. Further, the present inventors found no other relevant R228060 hydrochloride polymorph or patent literature. Accordingly, the present need in the art to develop a comprehensive system R228060 hydrochloride polymorph, found to be suitable to the development of crystalline form. The present inventors after many experiments, found that polymorph CS1 R228060 hydrochloride CS2 and a melting point polymorph, Form CS1 and CS2 is Form 183 ℃, much higher than the melting point disclosed in prior art solid. It provides a better alternative preparation of pharmaceutical preparations containing R228060 is, has very important implications for drug development.
PATENT
WO 2019027941
(i?)-2-amino-3-phenylpropyl carbamate (APC) is a phenylalanine analog that has been demonstrated to be useful in the treatment of a variety of disorders, including excessive daytime sleepiness, cataplexy, narcolepsy, fatigue, depression, bipolar disorder, fibromyalgia, and others. See, for example, US Patent Nos. 8,232,315; 8,440,715; 8,552,060; 8,623,913; 8,729,120; 8,741,950; 8,895,609; 8,927,602; 9,226,910; and 9,359,290; and U.S. Publication Nos. 2012/0004300 and 2015/0018414. Methods for producing APC (which also has other names) and related compounds can be found in US Patent Nos. 5,955,499; 5,705,640; 6,140,532 and 5,756,817. All of the above patents and applications are hereby incorporated by reference in their entireties for all purposes.
EXAMPLE 1
Synthesis of Compounds
Compound 8 (110CR002)
1 B 110CR002
[0083] tert- utyl (if)-(l-(Carbamothioyloxy)-3-phenylpropan-2-yl)carbamate (IB): A
60% dispersion of sodium hydride (0.36 g, 4.78 mmol, 1.2 equiv) in mineral oil was added in portions to compound 1A (1.0 g. 3.98 mmol, 1 equiv) in THF (20 mL) at 0 °C. After stirring for 1 hour, carbon disulfide (0.191 g, 4.78 mmol, 1.2 equiv) was added at 0 °C. After an additional hour of stirring, methyl iodide (0.3 mL, 4.78 mmol, 1.2 equiv) was added and the reaction was warmed to room temperature. After stirring two additional hours, concentrated ammonium hydroxide (1.6 mL, 7.98 mmol, 2 equiv) was added and the reaction was stirred overnight at room temperature. The reaction was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude compound IB. The solid was triturated in diethyl ether (20 mL) to give compound IB (0.17 g, 14% yield) as a light yellow solid.
[0084] (R)-0-(2-Amino-3-phenylpropyl) carbamothioate dihydrochloride (110CR002):
4M HCI in dioxane (0.68 mL, 2.74 mmol, 5 equiv) was added to neat compound IB (0.17 g, 0.548 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR003 (140 mg, 93% yield, 96.9% purity) as a white solid.
Compound 9 (110CR003)
Scheme 2
2A 2B 110CR003
[0085] (R)-2-((ter^Butoxycarbonyl)amino)-3-phenylpropyl sulfamate (2B): A solution of sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of compound 2 A (1.0 g, 3.98 mmol, 1 equiv) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for 4 hours, additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C. The reaction was stirred at room temperature overnight, at which point LCMS indicated a 3 :2 mixture of product to starting material. Additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C and the reaction was stirred at room temperature for an additional 6 hours. LCMS indicated a 4: 1 mixture of product to starting material. The reaction was quenched with saturated sodium bicarbonate (5 mL) and stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The product still contained unreacted starting material which could not be easily separated. Sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of crude compound 2B (0.9 g) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for two hours, the reaction was quenched with saturated sodium bicarbonate (5 mL) and the reaction was stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (Redisep 24 g silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 2B (0.37 g, 28% yield) as a white solid.
[0086] (R)-2-Amino-3-phenylpropyl sulfamate hydrochloride (110CR003): 4M HC1 in dioxane (1.4 mL, 5.6 mmol, 5 equiv) was added to neat compound 2B (0.37 g, 1.12 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under a vacuum at room temperature for two hours to give compound 110CR003 (250 mg, 84% yield, 97.8% purity) as a white solid.
Com ound 3 (110CR007)
[0087] (Benzyl (R)-(l-phenyl-3-ureidopropan-2-yl)carbamate) (3B): Concentrated hydrochloric acid (0.06 mL, 0.68 mmol, 0.12 equiv) was added to a solution of benzyl (ft)-(l -amino-3-phenylpropan-2-yl)carbamate ( 1.5 g, 5.28 mmol, 1 equiv) and urea (1.26 g, 21.21 mmol, 4 equiv) in toluene (150 mL) under nitrogen. After refluxing overnight, LCMS indicated the reaction was complete. The reaction was concentrated under reduced pressure, diluted with water (150 mL) and stirred for 30 minutes. The resulting solid was filtered and washed with water (25 mL) to give crude compound 3B (1.4 g, 4.27 mmol, 80% yield) as a white solid, which was used sequentially.
[0088] ((R)-l-(2-mino-3-phenylpropyl)urea) (3C): Compound 3B (0.5 g, 1.5 mmol, 1 equiv) and 10% palladium on carbon (0.09 g) in methanol (60 mL) was hydrogenated at 30 psi for 1 hour at which time LC-MS determined that the reaction was incomplete. The solution was filtered and fresh catalyst (0.09 g) was added. The solution was hydrogenated at 30 psi for an additional 45 minutes resulting in complete conversion. Two identical scale reactions were run for 105 minutes each, both resulting in complete conversion. The three runs were combined and filtered through celite, which was washed with methanol (50 mL). The filtrate was concentrated under reduced pressure to give crude compound 3C (0.9 g), which was used sequentially.
[0089] (R)-l-(2-Amino-3-phenylpropyI)urea hydrochloride (110CR007): Compound 3C (0.88 g, 4.58 mmol, 1 equiv) was dissolved diethyl ether (10 mL) and 4 N HCl in dioxane (2.31 mL, 9.27 mmol, 2 equiv) was added. The reaction was stirred overnight and then concentrated under reduced pressure to give crude 110CR007 as a white solid. The material was twice recrystallized from 10% methanol in ethanol (30 mL) to give 110CR007 (0.163 g, 16 % yield, 93.7 % purity) as a white solid.
Compound 4 (110CR009)
Scheme 4
[0090] Ethyl (R^)-4-((tert-butoxycarbonyI)amino)-5-phenylpent-2-enoate (4B): A solution of compound 4A (4.0 g, 16.1 mmol, 1 equiv) and ethyl (triphenylphos-phoranylidene)acetate (5.6 g, 16.1 mmol, 1 equiv) in dichloromethane (40 mL) was stirred at room temperature overnight. The reaction was concentrated under reduce pressure to remove the organic solvent and the resulting residue was purified on an AnaLogix automated system (40 g Sorbtech silica gel column), eluting with gradient of 50 to 100% ethyl acetate in heptanes, to give compound 4B (4.8 g, 94% yield) as a white solid.
[0091] (R^E)-4-((te *i-ButoxycarbonyI)amino)-5-phenylpent-2-enoic acid (4C): Lithium hydroxide (1.4 g, 60 mmol, 4 equiv) in water (15 mL) was added to compound 4B (4.8 g, 15 mmol, 1 equiv) in THF (60 mL) at room temperature and the reaction was stirred overnight. After 16 hours, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers was washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4C (4.2 g, 97% yield) as a light cream solid, which was used subsequently.
[0092] Methyl (R E)-4-((½ -i-butoxycarbonyl)amino)-5-phenylpent-2-enoate (4D1):
Isobutyl chloro formate (1.3 mL, 10 mmol, 1 equiv) in THF (4 mL) was added dropwise to a solution of compound 4C (3.0 g, 10 mmol, 1 equiv) and N-methyl-morpholine (1.1 mL, 10 mmol, 1 equiv) in THF (12 mL) at -15 °C. After 30 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 2M Ammonia in methanol (5 mL, 10 mmol, 1 equiv) was added dropwise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring, the reaction was warmed to room
temperature and stirred overnight. The reaction mixture was concentrated at reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (50 mL) and washed with water (100 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (80 g Sorbtech silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 4D1 (1.1 g, 35 % yield) as a white solid.
[0093] Methyl (S)-4-((te^-butoxycarbonyl)amino)-5-phenylpentanoate (4D2): A mixture of compound 4D1 (1.1 g, 3.6 mmol, 1 equiv) and 10% palladium on carbon (0.33 g, 50% wet) in methanol (40 mL) was hydrogenated at 40 psi at room temperature for 4 hours. The mixture was filtered through celite, which was washed with methanol (100 mL). The filtrate was concentrated under reduced pressure to give compound 4D2 (1.1 g, 99% yield) as a white solid.
[0094] (S)-4-((ii? i-Butoxycarbonyl)amino)-5-phenylpentanoic acid (4D3): Lithium hydroxide (73 mg, 3 mmol, 1.5 equiv) in water (1 mL) was added to compound 4B (0.6 g, 2 mmol, 1 equiv) in THF (9 mL) at room temperature. After stirring overnight, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (3 x 25 mL). The combined organic layers was washed with saturated brine (25 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4D3 (0.56 g, 98% yield) as a white solid, which was used subsequently.
[0095] tert-Butyl (S)-(5-amino-5-oxo-l-phenylpentan-2-yl)carbamate (4E): Isobutyl chloroformate (0.23 mL, 1.8 mmol, 1 equiv) in THF (0.5 mL) was added drop-wise to a solution of compound 4C (0.54 g, 1.8 mmol, 1 equiv) and N-methylmorpholine (0.2 mL, 1.8 mmol, 1 equiv) in THF (1 mL) at -15 °C. After 20 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 0.4M Ammonia in THF (9 mL, 3.6 mmol, 2 equiv) was added drop-wise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring the reaction was warmed to room temperature and stirred overnight. The reaction mixture was concentrated under reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (25 mL) and washed with water (25 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 x 25 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under
reduced pressure to give compound 4E (0.5 g, 93% yield) as a white solid, which was used subsequently.
[0096] (S)-4-Amino-5-phenylpentanamide hydrochloride (110CR009): 4M HC1 in dioxane (6 mL, 25 mmol, 10 equiv) was added to compound 4E (0.73 g, 1.12 mmol, 1 equiv) After stirring overnight at room temperature, the reaction was diluted with diethyl ether (20 mL) and stirred for 6 hours. The resulting suspension was filtered and the solid was washed with diethyl ether (20 mL). The filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR009 (340 mg, 60% yield, 97.9 % purity) as a white solid.
Compound 10 (110CR012)
[0097] tert-Butyl (R)-(l-(carbamoylthio)-3-phenyIpropan-2-yI)carbamate (5B):
Compound 5 A (0.15 g, 0.56 mmol, 1 equiv) was dissolved in THF (8 mL) and sparged with nitrogen for 15 minutes. Trichloroacetyl isocyanate (0.1 mL, 0.84 mmol, 1.5 equiv) was added and the solution stirred for 3 hours, at which point TLC (30% ethyl acetate in heptane) indicated absence of starting material. The reaction was cooled to 0°C and concentrated ammonium hydroxide (0.15 mL) was added. After stirring overnight at room temperature, TLC indicated that the reaction was complete. The reaction was washed with a 10% ammonium hydroxide (10 mL). The organic layer was concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (12 g silica gel column), eluting with a gradient of 0 to 30% ethyl acetate in heptane, to give compound 5B. This reaction was repeated an additional two times 0.15 g and 0.18 g). The products were to give compound 5B (0.35 g, 1.12 mmol, 62.2% yield) as a white solid.
[0098] (R)-S-(2-Amino-3-phenylpropyl) carbamothioate hydrochloride (110CR012):
Compound 5B (0.35 g, 1.12 mmol, 1 equiv) was dissolved in 4N HCI in dioxane (2 mL). The reaction was stirred for two hours and then concentrated under reduced pressure to give crude 110CR012 as a white solid. The material was triturated in diethyl ether (15 mL) to give 110CR012 (0.215 g, 78 % yield, 98.0 % purity) as a white solid.
References
- ^ Jump up to:a b “SUNOSI
![™]() (solriamfetol) Tablets, for Oral Use. Full Prescribing Information” (PDF). Jazz Pharmaceuticals. 2019. Retrieved 21 March2019.
(solriamfetol) Tablets, for Oral Use. Full Prescribing Information” (PDF). Jazz Pharmaceuticals. 2019. Retrieved 21 March2019. - ^ Abad, VC; Guilleminault, C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC 5344488. PMID 28424564.
- ^ Jump up to:a b c d Ji-young, Sohn (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan, SS; Guilleminault, C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID 26558298.
- ^ Garde, Damian (January 14, 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:a b “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ de Biase, S; Nilo, A; Gigli, GL; Valente, M (August 2017). “Investigational therapies for the treatment of narcolepsy”. Expert Opinion on Investigational Drugs. 26 (8): 953–963. doi:10.1080/13543784.2017.1356819. PMID 28726523.
 (solriamfetol) Tablets, for Oral Use. Full Prescribing Information”
(solriamfetol) Tablets, for Oral Use. Full Prescribing Information”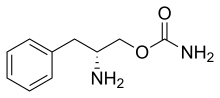 |
|
| Clinical data | |
|---|---|
| Trade names | Sunosi |
| Synonyms | SKL-N05, ADX-N05, ARL-N05, and JZP-110; (R)-2-amino-3-phenylpropylcarbamate hydrochloride |
| Routes of administration |
By mouth |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | ~95% |
| Protein binding | 13.3–19.4% |
| Metabolism | negligible |
| Elimination half-life | ~7.1 h |
| Excretion | urine (95% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C10H14N2O2 |
| Molar mass | 194.234 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////Solriamfetol hydrochloride, Solriamfetol, ソルリアムフェトル塩酸塩; солриамфетол , سولريامفيتول , 索安非托 , JZP-110, Orphan Drug, fda 2019, R228060, R 228060