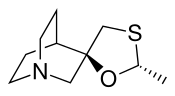
Cevimeline
セビメリン
- Molecular FormulaC10H17NOS
- Average mass199.313 Da
Cevimeline hydrochloride
-
- Synonyms:AF-102B, SNI-2011, SNK-508, Evoxac
- ATC:N07
- Use:cognition disorder, treatment of Sjogren’s syndrome, muscarinic M3-receptor agonist
- Chemical name:(2′R,3R)-rel-2′-methylspiro[1-azabicyclo[2.2.2]octane-3,5′-[1,3]oxathiolane] hydrochloride hydrate (2:2:1)
- Formula:C10H17NOS • HCl • 1/2H2O
- MW:489.57 g/mol
- CAS-RN:153504-70-2
- InChI Key:SURWTGAXEIEOGY-GHXDPTCOSA-N
- InChI:InChI=1S/C10H17NOS.ClH/c1-8-12-10(7-13-8)6-11-4-2-9(10)3-5-11;/h8-9H,2-7H2,1H3;1H/t8-,10-;/m1./s1
Derivatives
base
- Formula:C10H17NOS
- MW:199.32 g/mol
- CAS-RN:107233-08-9
anhydrous hydrochloride
- Formula:C10H17NOS • HCl
- MW:235.78 g/mol
- CAS-RN:107220-28-0
Cevimeline is cis-2′-methylspiro {1-azabicyclo [2.2.2] octane-3, 5′ -[1,3] oxathiolane} hydro-chloride, hydrate (2:1). Its empirical formula is C10H17NOS•HCl•½ H2O, and its structural formula is:
 Cevimeline has a molecular weight of 244.79. It is a white to off white crystalline powder with a melting point range of 201 to 203°C. It is freely soluble in alcohol and chloroform, very soluble in water, and virtually insoluble in ether. The pH of a 1% solution ranges from 4.6 to 5.6. Inactive ingredients include lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate.
Cevimeline has a molecular weight of 244.79. It is a white to off white crystalline powder with a melting point range of 201 to 203°C. It is freely soluble in alcohol and chloroform, very soluble in water, and virtually insoluble in ether. The pH of a 1% solution ranges from 4.6 to 5.6. Inactive ingredients include lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate.
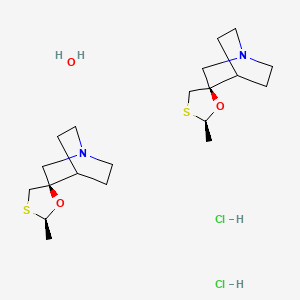
Cevimeline hydrochloride [USAN]
RN: 153504-70-2
(+-)-cis-2-Methylspiro(1,3-oxathiolane-5,3′-quinuclidine) hydrochloride, hemihydrate
Cevimeline (trade name Evoxac) is a parasympathomimetic and muscarinic agonist,[1] with particular effect on M1 and M3 receptors. It is used in the treatment of dry mouth and especially associated with Sjögren’s syndrome.
Mechanism of action
By activating the M3 receptors of the parasympathetic nervous system, cevimeline stimulates secretion by the salivary glands, thereby alleviating dry mouth.
Side effects
Known side effects include nausea, vomiting, diarrhea, excessive sweating, rash, headache, runny nose, cough, drowsiness, hot flashes, blurred vision, and difficulty sleeping.[2]
Contraindications include asthma and angle closure glaucoma.
Clip
https://www.sciencedirect.com/science/article/abs/pii/S0731708515302260



CLIP
https://www.sciencedirect.com/science/article/pii/S0040403913005042



CLIP
CLIP

- Reaction of quinuclidin-3-one (I) with trimethylsulfoxonium iodide and NaH in DMSO gives epoxide (II), which is opened with SH2 in NaOH/water, yielding 3-hydroxy-3-(sulfanylmethyl)quinuclidine (III). The cyclization of compound (III) with acetaldehyde (IV) catalyzed by boron trifluoride ethearate or by SnCl4, POCl3, H3PO4 or p-toluenesulfonic acid affords a mixture of two diastereomeric spiroracemates, the (?-trans (V) and (?-cis (cevimeline). This mixture is separated by fractional recrystallization in acetone or by TLC chromatography, and treated with hydrochloric acid. The (?-trans-compound (V) can be isomerized to cevimeline by treatment with an acidic catalyst such as an organic sulfonic acid (trifluoromethanesulfonic acid, p-toluenesulfonic acid or methanesulfonic acid), a Lewis acid (SnCl4, FeCl3, BF3 or AlCl3) or sulfuric acid in refluxing toluene, hexane or CHCl3. Cevimeline hydrochloride hemihydrate is obtained from the above mentioned hydrochloride by a complex work-up using water, isopropanol and n-hexane.
- Synthesis of Cevimeline Hydrochloride (EN:134916): Reaction of quinuclidin-3-one (I) with trimethylsulfoxonium iodide and NaH in DMSO gives epoxide (II), which is opened with SH2 in NaOH/water, yielding 3-hydroxy-3-(sulfanylmethyl)quinuclidine (III) (1,2). The cyclization of compound (III) with acetaldehyde (IV) catalyzed by boron trifluoride ethearate (1) or by SnCl4, POCl3, H3PO4 or p-toluenesulfonic acid (2) affords a mixture of two diastereomeric spiro-racemates, the (?-trans (V) and (?-cis (cevimeline). This mixture is separated by fractional recrystallization in acetone or by TLC chromatography, and treated with hydrochloric acid (1,2). The (?-trans-compound (V) can be isomerized to cevimeline by treatment with an acidic catalyst such as an organic sulfonic acid (trifluoromethanesulfonic acid, p-toluenesulfonic acid or methanesulfonic acid), a Lewis acid (SnCl4, FeCl3, BF3 or AlCl3) or sulfuric acid in refluxing toluene, hexane or CHCl3 (2,3). Cevimeline hydrochloride hemihydrate is obtained from the above mentioned hydrochloride by a complex work-up using water, isopropanol and n-hexane (4).(Scheme 13491601a) Description M.p. 203 C (4). Sources Discovered by Israel Institute for Biological Research, Ness-Ziona (IL) and licensed to Snow Brand Milk Products Co. Ltd. (JP). In the U.S., comarketed by Snow Brand Milk Products and Daiichi Pharmaceutical Co., Ltd. In Japan, codeveloped with Nippon Kayaku Co. Ltd. Ishihara Sangyo Co., Ltd. (JP) is the bulk supplier. References 1. Fisher, A., Heldman, E., Grunfeld, Y., Karton, I., Levy, A. (Israel Institute for Biological Research); Derivs. of quinuclidine; EP 0205247, JP 1986280497, US 4855290. 2. Hayashi, K., Tokumoto, S., Yoshizawa, H., Isogai, T. (Ishihara Sangyo Kaisha, Ltd.); Method for producing 2-methylspiro(1,3-oxathiolan-5,3′)quinuclidine; EP 0683168, US 5571918. 3. Haga, T., Koyanagi, T., Hara, K., Maeda, M., Shigehara, I. (Ishihara Sangyo Kaisha, Ltd.); Method for isomerization of trans-form 2-methylspiro(1,3-oxathiolane-5,3′)quinuclidine or acid addition salts thereof; EP 0298491, US 4861886. 4. Saito, K., Ono, T., Honda, N. (Snow Brand Milk Products Co., Ltd.); Preparation method of cis-2-methylspiro(1,3-oxathiolane-5,3′)quinuclidine hydrochloride.1/2 hydrate capable of disgregating easily; JP 1992108792.
PATENT
https://patents.google.com/patent/US8080663B2/en
The present invention refers to a novel, industrially advantageous process for the preparation of an intermediate useful for the preparation of Cevimeline hydrochloride (1, cis-2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine, Scheme 1). This pharmaceutical is useful for the treatment of diseases of the central nervous system due to disturbances of central cholinergic function and autoimmune system (Sjörgen’s syndrome) and is marketed as Evoxac®.
U.S. Pat. No. 4,855,290 describes a process for preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1). The process comprises the preparation of the epoxide of 3-methylenequiniclidine, which is subsequently reacted with hydrogen sulfide to produce 3-hydroxy-3-mercaptomethylquiniclidine and condensed with acetaldehyde in the presence of a Lewis acid (boron trifluoride etherate) to provide 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine. This process is depicted in Scheme I.

This process suffers from major disadvantages when transiting to industrial scale. These include the use of the highly hazardous and difficult to handle hydrogen sulfide gas. Also, boron trifluoride etherate is employed during the condensation step with acetaldehyde. The boron trifluoride etherate reagent is an air and moisture sensitive Lewis acid which has to be used under anhydrous conditions, thus creating a serious disadvantage in industrial settings. Another drawback of this process is the use of sodium hydride. U.S. Pat. Nos. 5,571,918 and 4,861,886 relate to the isomerization of the trans- to cis-form of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine but do not describe methods for its preparation. Thus, an industrially acceptable and cost-effective method for the preparation of Cevimeline hydrochloride which overcomes the deficiencies of the prior art is required.
Further and other objects of the invention will be realized by those skilled in the art from the following Summary of the Invention and Detailed Description of Preferred Embodiments of the Invention thereof.
According to one aspect of the invention, a novel process is provided for the preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1). The process is industrially practical, efficient, safe and economical, as well as being environmentally friendly. The general method is shown in the Scheme II.

wherein R is selected from C1 to C6 alkyl and aryl groups, most preferably a methyl, ethyl or propyl group; R1 is hydrogen or a C2 to C7 alkyl or aryl carbonyl group; R2 is a C1 to C6 alkyl group, preferably methyl, ethyl, propyl, or butyl group.
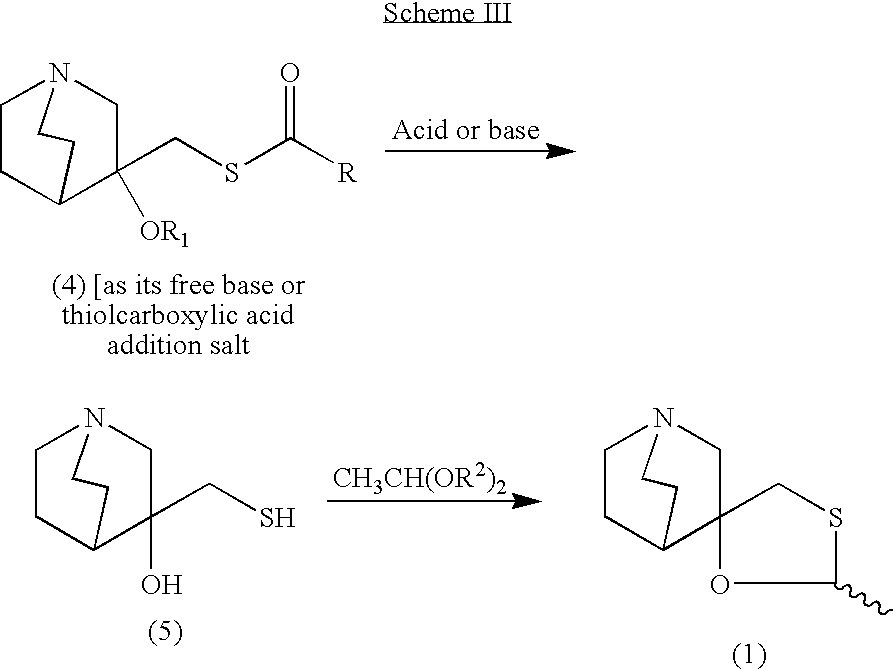
EXAMPLE I Preparation of the Epoxide of 3-methylenequiniclidine (3)
A mixture of the hydrochloric salt of 3-quiniclidinone (2, 120 g, 795.7 mmol) and trimethylsulfoxonium iodide (219 g, 993.3 mmol) in dimethylsulfoxide (91.0 g, 0.63 mol) was cooled to 0-5° C. in an ice/water bath under nitrogen atmosphere. A solution of potassium tert-butoxide (201 g, 1789.1 mmol) in dimethylsulfoxide (500 mL) was added dropwise over 45 minutes. The mixture was warmed gradually to room temperature and stirred for an additional 16 hours at room temperature. After cooling to 0-5° C. (ice/water bath) the mixture was poured into an ice/water mixture (500 g) and then sodium chloride (300 g) was added. The mixture was stirred for 30 minutes and extracted with toluene (3×400 mL). The toluene phase was dried over sodium sulfate, filtered and evaporated to furnish the epoxide of 3-methylenequiniclidine (60 g, 431.7 mmol, 54% yield) as a yellow oil. The product could be used in the next step neat or as toluene solution after the extraction without further purification.
1H NMR (400 MHz, CDCl3): δ=3.10 (d, 1H, J=14.6 Hz); 2.98-2.77 (m, 5H); 2.74 (d, 1H, J=4.8 Hz); 2.70 (d, 1H, J=4.8 Hz); 1.96-1.89 (m, 1H); 1.79-1.62 (m, 2H); 1.60-1.54 (m, 1H); 1.38-1.36 (m,1H).
LRMS (ES+): 140.0 (100, M+H+).
EXAMPLE II Preparation of the Thiolacetic Acid Salt of 3-hydroxy-3-acetoxymercaptomethylquiniclidine (4)
A solution of the epoxide of 3-methylenequiniclidine (3, 54 g, 388.5 mmol) in toluene (200 mL) was cooled to 0-5° C. (ice/water bath). Thiolacetic acid was added dropwise over 10-15 minutes. The mixture was stirred at 0-5° C. for 30 minutes and then allowed to come to room temperature. After stirring at room temperature for 2 hours the formed precipitate was filtered and washed with toluene (2×100 mL) to give the 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 77 g, 264.6 mmol, 68%) as a light yellow solid. The product was used in the next step without any further purification.
1H NMR (400 MHz CD3OD): δ=3.47 (d, 1H, J=14.1 Hz); 3.37-3.18 (m, 7H); 2.40 (s, 3H); 2.38 (s, 3H); 2.36-2.27 (m, 1H), 2.14-2.05 (m, 2H); 2.03-1.93 (m, 1H); 1.81-1.78 (m, 1H).
LRMS (ES+): 216.1 (100, M−[SCOCH3]−+H+).
EXAMPLE III Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine using p-toluenesulfonic acid (1)
To a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL) was added p-toluenesulfonic acid monohydrate (5.9 g, 30.9 mmol) and the mixture was heated to reflux for 3.5 hours. The mixture was cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was heated to reflux and stirred for an additional 3 hours. The solvent was evaporated and the residue was dissolved in dichloromethane (50 mL). The mixture was cooled to 0-5° C. and a 25% aqueous solution of sodium hydroxide (80 mL) was added. The mixture was stirred for 10-15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (3×50 mL). The organic phases were combined and extracted with 5% aqueous solution of sulfuric acid (3×50 mL). The acidic aqueous phases were combined and the pH was adjusted to 12 with a 25% aqueous solution of sodium hydroxide. The aqueous phase was extracted with heptane (3×50 mL) and the organic phases were combined, dried over sodium sulfate and the solvent was evaporated to give 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.8 g, 9.2 mmol, 89% yield) as a 3:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
LRMS (ES+): 200.1 (100, M+H+).
EXAMPLE IV Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using racemic camphorsulfonic acid
In a similar experiment as Example III, racemic camphorsulfonic acid (7.2 g, 30.9 mmol) was added to a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL). The mixture was refluxed for 5 h, cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was refluxed for an additional an 8 hours and processed according to Example III to give 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.32 g, 6.63 mmol, 64% yield) in a 3.5:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
EXAMPLE V Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using phenyl sulfonic acid
In a similar experiment as Example III, to a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL) was added phenyl sulfonic acid (4.9 g, 30.9 mmol) and the mixture was refluxed 5 h, cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was refluxed for an additional 8 hours and worked up in a manner similar to Example III to furnish 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.6 g, 8.2 mmol, 80% yield) as a 2.5:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
EXAMPLE VI Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using p-toluenesulfonic acid in butanol
To a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in butanol (100 mL) was added of p-toluenesulfonic acid monohydrate (5.9 g, 30.9 mmol) and the mixture was refluxed for 3 hours with a Dean-Stark apparatus attached to the flask. The reaction mixture was cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was heated to 80° C. for an additional 8 h and worked up according to Example III to afford 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.8 g, 9.2 mmol, 89% yield) as a 3:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
References
- ^ Ono M, Takamura E, Shinozaki K, et al. (July 2004). “Therapeutic effect of cevimeline on dry eye in patients with Sjögren’s syndrome: a randomized, double-blind clinical study”. Am. J. Ophthalmol. 138 (1): 6–17. doi:10.1016/j.ajo.2004.02.010. PMID 15234277.
- ^ [1] MedicineNet: Cevimeline. Accessed 10/12/2007
-
- US 4 855 290 (Israel Institute for Biological Research; 8.8.1989; IL-prior. 10.5.1985).
- US 4 876 260 (Israel Institute for Biological Research; 24.10.1989; USA-prior. 28.10.1987).
- EP 683 168 (Ishihara Sangyo Kaisha; appl. 19.5.1995; J-prior. 19.5.1994).
-
Method for isomerization of trans-isomer:
- US 4 861 886 (Ishihara Sangyo Kaisha; 29.8.1989; J-prior. 10.7.1987).
-
Method of separation:
- IL 81 652 (Israel Institute for Biological Research; 12.5.1991; appl. 23.2.1987).
- JP 01 290 680 (Ishihara Sangyo Kaisha; 22.11.1989; J-prior. 18.5.1988).
-
Synthesis of enantiomerically pure (S)-3-hydroxy-3-mercaptomethylquinuclidine (S)-II:
- Bos, M.; Canesso, R.: Heterocycles (HTCYAM) 38 (8), 1889 (1994).
-
Synthesis of 3-quinuclidone:
- Sternbach, L.H.; Kaiser, S.: J. Am. Chem. Soc. (JACSAT) 74, 2215 (1952).
-
External links
|
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Evoxac |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608025 |
| Pregnancy category |
|
| Routes of administration |
By mouth (capsules) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | <20% |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C10H17NOS |
| Molar mass | 199.31308 g/mol g·mol−1 |
| 3D model (JSmol) | |


/////////// Cevimeline, AF-102B, SNI-2011, SNK-508, Evoxac, セビメリン