
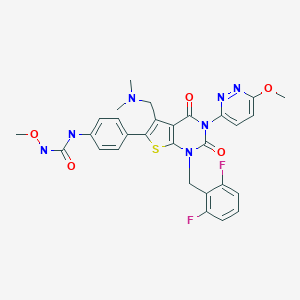
Relugolix (TAK-385), RVT 601
レルゴリクス
| Formula |
C29H27F2N7O5S
|
|---|---|
| CAS |
737789-87-6
|
| Mol weight
UNII |
623.6304
UNII-P76B05O5V6
|
2019/1/8 PMDA JAPAN APPROVED, Relumina

- Originator Takeda
- Developer Myovant Sciences; Takeda; Takeda Oncology
- Class Analgesics; Antineoplastics; Ketones; Pyrimidines; Small molecules
- Mechanism of Action LHRH receptor antagonists
- Preregistration Uterine leiomyoma
- Phase III Pain; Prostate cancer
- No development reported Solid tumours
- 08 Nov 2018 Myovant announces intention to submit NDA for Uterine leiomyoma in Q3 of 2019
- 08 Nov 2018 Myovant Sciences completes enrollment in the phase III LIBERTY 1 trial for Uterine leiomyoma (Combination therapy) in USA (PO)(NCT03049735)
- 25 Oct 2018 Myovant Sciences completes enrolment in its phase III HERO trial for Prostate cancer (Late-stage disease) in Denmark, Australia, Austria, Belgium, Canada, United Kingdom, USA, Japan, Taiwan, Sweden, Spain, Slovakia, New Zealand, Netherlands, South Korea, Germany, France and Finland (PO) (NCT03085095)

Relugolix has been used in trials studying the treatment of Endometriosis, Prostate Cancer, Uterine Fibroids, and Androgen Deprivation Treatment-naïve Nonmetastatic Prostate Cancer.
Relugolix (developmental code names RVT-601, TAK-385) is a gonadotropin-releasing hormone antagonist (GnRH antagonist) medication which is under development by Myovant Sciences and Takeda for the treatment of endometriosis, uterine fibroids, and prostate cancer.[1][2][3][4][5][6][7] Unlike most other GnRH modulators, but similarly to elagolix, relugolix is a non-peptide and small-molecule compound and is orally active.[6][7] As of July 2018, it is in the pre-registration phase of development for uterine fibroids and is in phase III clinical trials for endometriosis and prostate cancer.[1]
Pharmacology
Pharmacodynamics
Relugolix is a selective antagonist of the gonadotropin-releasing hormone receptor (GnRHR) (IC50 = 0.12 nM).[6][7][8]
A single oral administration of relugolix at a dose of 3 mg/kg has been found to suppress luteinizing hormone (LH) levels for more than 24 hours in castrated cynomolgus monkeys, indicating a long duration of action.[6] The drug (80–160 mg/day) has been found to reduce testosterone levels to sustained castrate levels in men with once-daily administration.[8] Lower dosages (10–40 mg/day) are being studied in the treatment of endometriosis and uterine fibroids to achieve partial sex hormone suppression.[4] The reasoning behind partial suppression for these conditions is to reduce the incidence and severity of menopausal symptoms such as hot flushes and to avoid bone mineral density changes caused by estrogen deficiency that can eventually lead to osteoporosis.[4][9]
History
Relugolix was first described in 2004.[10][6] It superseded sufugolix, which was developed by the same group.[6]
Society and culture
Generic names
Relugolix is the generic name of the drug and its INN and USAN.[11] It is also known by its developmental code names RVT-601 and TAK-385.[1][11]
PATENT
http://www.google.co.in/patents/EP1591446A1?cl=en
(Production Method 1)

- (Production method 2)
-
![Figure 00130001]()

-
-
- Example 83
-
http://www.google.co.in/patents/EP1591446A1?cl=en
- Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
-
![Figure 01690002]()
-
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals.
1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).

PAPER
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (16b)
 tak 385
tak 385
http://pubs.acs.org/doi/suppl/10.1021/jm200216q/suppl_file/jm200216q_si_001.pdf
PATENT
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
PATENT
https://patents.google.com/patent/WO2015062391A1/en
Endometriosis is a common estrogen-dependent gynecological diseases, often occurs in women during their childbearing years, and its mechanism is unclear. Complex and difficult to diagnose the cause of the symptoms of endometriosis is unknown, serious block to the discovery of effective therapies. Currently, endometriosis primarily by laparoscopy diagnosis, and treatment by surgery, or pill, or progesterone receptor agonists of GnRH reduce estrogen levels to control.
Currently the high incidence of endometriosis, Datamonitor 2009 year data show that only two countries, India and China, the number of female patients suffering from endometriosis had more than 68 million (31,288,000 India, China 3753.5 million) passengers, while the national prevalence of the number seven major markets have more than 17 million. Datamonitor expects 2009 to 2018, endometriosis market from 2009 to $ 764 million (US $ 596 billion and the EU $ 117 million, Japan US $ 051 million) in 2018 increased to US $ 1.156 billion (US 8.44 billion dollars, 206 million US dollars the European Union, Japan $ 106 million), while the Chinese market will have more room for growth.
Gonadotropin-releasing hormone (Gonadoliberin; gonadotropin releasing hormone; GnRH), also known as luteinizing hormone releasing hormone (LHRH), is synthesized by neuroendocrine cells of the hypothalamus hormones decapeptide (pGlu-His-Trp-Ser-Tyr-Gly- Leu-Arg-Pro-Gly-NH2), a central regulator of reproductive endocrine system. Which conveys the circulatory system through hypothalamus-pituitary portal to the pituitary, bind to the cells of the anterior pituitary GnRH receptor, such as gonadotropin luteinizing hormone (Luteinizing Hormone, LH) and FSH (Follicle-Stimulating Hormone, FSH ) secretion and release, regulation of normal development and corpus luteum of the ovary, hypothalamic – pituitary – gonadal axis plays an important role. GnRH receptors capable of activating the G protein coupled calcium phosphatidylinositol second messenger system exert their regulatory role, and LH is adjusted to produce steroids, FSH regulating development of the male and female follicle spermatogenesis.
LH and FSH are released into the circulation, and combined with the ovaries or testes specific cell receptors, stimulating the production of steroids. The presence of sex steroids, diseases such as endometriosis, uterine fibroids, prostate cancer and exacerbations, to be given long-acting GnRH receptor agonists and antagonists for treatment control peptides.
Peptide GnRH receptor antagonists include linear peptides (US 5,171,835) GnRH-derived, cyclic hexapeptide derivatives (US 2002/0065309), a bicyclic peptide derivative (Journal of Medicinal Chemistry, 1993; 36: 3265-73), etc. ; and GnRH receptor peptide agonists include leuprolide (leuprorelin, pGlu-His-Trp-Ser-Tyr-d-Leu-Leu-Arg-Pro-NHEt). However, there are many problems including oral absorbability, dosage form, dose volume, drug stability, sustained action, and metabolic stability of the peptide-type compound to be resolved. But the main reason small molecule GnRH receptor antagonists of peptide-based therapy is superior to the existing method is that small molecule GnRH receptor antagonist may be orally administered directly, convenient. Studies have shown that small molecule antagonists of endometriosis, precocious puberty, prostate cancer and other hormone-dependent diseases having a significant effect.
GnRH receptor agonist mediated indirect mechanisms of tumor suppression by long-term effects on the hypothalamic – pituitary – gonadal axis, leading to pituitary gonadotropins (FSH, LH) is reduced, thereby reducing the secretion of sex hormones and indirectly inhibit growth of tumor cells. And a GnRH receptor antagonist directly to inhibit the release of the pituitary gonadotropins, thereby inhibiting tumor cell growth.
Given the limitations of peptide GnRH receptor antagonists, non-peptide GnRH receptor antagonists have been proposed and into the development, clinical trials and launch phase, such as Elagolix (NBI-56418, or also known as ABT-620) is a Abbott and Neurocrine Biosciences Inc company co-developed small molecule GnRH receptor antagonist, is currently in phase III clinical stage, mainly used in the treatment of endometriosis (III phase) and uterine fibroids (II period). June 2012, data released results of a Phase II clinical endometrial endometriosis Houston, the 94th annual meeting of the Endocrine Society: 131 accepts elagolix (150 or 250mg qd), leuprorelin depot (3.75mg sc in, once a month, female patients with endometriosis endometrium 12 weeks) or placebo treatment, elagolix treatment groups in patients with serum hormone estrogen compared to leuprorelin therapy group and the placebo group was significantly reduced. At the same time, elagolix safety and tolerability have been well verified.
Relugolix also known as TAK-385, is a GnRH by the Japanese Takada Pharmaceutical company developed an oral small molecule receptor antagonist, for the treatment of endometriosis, uterine fibroids and prostate. 2011 entered endometriosis and uterine fibroids clinical phase II study, carried out a clinical study of prostate cancer in the same year.
It disclosed a series of current small molecule GnRH receptor antagonists including patent WO2006096785, WO2010026993, WO2011076687, WO2012175514 like.
Despite the large number of interesting studies have been conducted in this field, there remains a need to continue research and development of more effective small molecule GnRH receptor antagonists, the present invention provides a novel GnRH receptor antagonist structure, and found to have such a structure compounds having good activity, reproductive endocrine system effective to treat the disease.
Example 15
1- (4- (1- (2,6-difluorobenzyl) -5 – ((dimethylamino) methyl) -3- (2-fluoro-3-methoxyphenyl) -2,4 – dioxo-1,2,3,4-tetrahydro-pyrrolo [2,1-f] [1,2,4] triazin-6-yl) phenyl) -3-methoxy-urea


first step
1- (4- (1- (2,6-difluorobenzyl) -5 – ((dimethylamino) methyl) -3- (2-fluoro-3-methoxyphenyl) -2,4 – dioxo-1,2,3,4-tetrahydro-pyrrolo [2,1-f] [1,2,4] triazin-6-yl) phenyl) -3-methoxy-urea
6- (4-aminophenyl) -1- (2,6-difluorobenzyl) -5 – ((dimethylamino) methyl) -3- (2-fluoro-3-methoxyphenyl ) pyrrolo [2,1-f] [1,2,4] triazine -2,4 (1H, 3H) – dione 13j (55mg, 0.10mmol) was dissolved in 4mL of tetrahydrofuran, was added p-nitrophenyl chloroformate (30mg, 0.15mmol) and pyridine (32mg, 0.40mmol), the reaction 30 ℃ 3 h, methoxyamine hydrochloride (25mg, 0.30mmol), seal the reaction for 8 hours the reaction was stopped. Distillation under reduced pressure, the residue was purified by thin-layer chromatography using three developing solvent system A to give the title product 1- (4- (1- (2,6-difluorobenzyl) -5 – ((dimethylamino) methyl yl) -3- (2-fluoro-3-methoxyphenyl) -2,4-dioxo-1,2,3,4-tetrahydro-pyrrolo [2,1-f] [1,2 , 4] triazin-6-yl) phenyl) -3-methoxy-urea 15 (5mg, yellow solid), yield: 8.1%.
MS m / z (ESI): 623.4 [M + 1]
1 H NMR (400MHz, DMSO- d 6) 7.90-7.87 (m, 2H), 7.85-7.80 (m, 2H), 7.60 (s, 1H), 7.55-7.25 (m, 4H), 7.09-7.02 (m , 1H), 6.71-6.65 (m, 1H), 4.50 (s, 2H), 3.84 (s, 3H), 3.67 (s, 3H), 3.54 (s, 2H), 2.15 (s, 6H)
References
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
References
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800028257
- ^ Goenka L, George M, Sen M (June 2017). “A peek into the drug development scenario of endometriosis – A systematic review”. Biomed. Pharmacother. 90: 575–585. doi:10.1016/j.biopha.2017.03.092. PMID 28407578.
- ^ Dellis A, Papatsoris A (October 2017). “Therapeutic outcomes of the LHRH antagonists”. Expert Rev Pharmacoecon Outcomes Res. 17 (5): 481–488. doi:10.1080/14737167.2017.1375855. PMID 28870102.
- ^ Jump up to:a b c Streuli I, de Ziegler D, Borghese B, Santulli P, Batteux F, Chapron C (March 2012). “New treatment strategies and emerging drugs in endometriosis”. Expert Opin Emerg Drugs. doi:10.1517/14728214.2012.668885. PMID 22439891.
- ^ Elancheran, R.; Maruthanila, V. L.; Ramanathan, M.; Kabilan, S.; Devi, R.; Kunnumakara, A.; Kotoky, Jibon (2015). “Recent discoveries and developments of androgen receptor based therapy for prostate cancer”. Med. Chem. Commun. 6 (5): 746–768. doi:10.1039/C4MD00416G. ISSN 2040-2503.
- ^ Jump up to:a b c d e f Miwa K, Hitaka T, Imada T, Sasaki S, Yoshimatsu M, Kusaka M, Tanaka A, Nakata D, Furuya S, Endo S, Hamamura K, Kitazaki T (July 2011). “Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor”. J. Med. Chem. 54 (14): 4998–5012. doi:10.1021/jm200216q. PMID 21657270.
- ^ Jump up to:a b c Nakata D, Masaki T, Tanaka A, Yoshimatsu M, Akinaga Y, Asada M, Sasada R, Takeyama M, Miwa K, Watanabe T, Kusaka M (January 2014). “Suppression of the hypothalamic-pituitary-gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: studies in human GnRH receptor knock-in mice”. Eur. J. Pharmacol. 723: 167–74. doi:10.1016/j.ejphar.2013.12.001. PMID 24333551.
- ^ Jump up to:a b MacLean D, Shi H, Suri A, Faessel H, and Saad F (2013). “Safety and Testosterone-Lowering Effects of the Investigational, Oral, GnRH Antagonist, TAK-385 in Healthy Male Volunteers: Results of a Phase 1 Inpatient/Outpatient Study”. doi:10.1210/endo-meetings.2013.CN.1.SAT-318.
- ^ Struthers RS, Nicholls AJ, Grundy J, Chen T, Jimenez R, Yen SS, Bozigian HP (February 2009). “Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix”. J. Clin. Endocrinol. Metab. 94 (2): 545–51. doi:10.1210/jc.2008-1695. PMC 2646513. PMID 19033369.
- ^ https://patents.google.com/patent/US7300935/
- ^ Jump up to:a b https://chem.nlm.nih.gov/chemidplus/rn/737789-87-6
|
|
 |
|
| Clinical data | |
|---|---|
| Synonyms | RVT-601; TAK-385 |
| Routes of administration |
By mouth |
| Drug class | GnRH antagonist |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C29H27F2N7O5S |
| Molar mass | 623.630 g/mol |
| 3D model (JSmol) | |
External links
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 1 | Completed | Basic Science | Healthy Volunteers | 1 |
| 1 | Completed | Treatment | Androgen Deprivation Treatment-naïve Nonmetastatic Prostate Cancer / Hormone Treatment-naïve Participants With Prostate Cancer | 1 |
| 1 | Completed | Treatment | Endometriosis / Prostate Cancer | 1 |
| 1 | Completed | Treatment | Japanese Premenopausal Healthy Adult Women | 1 |
| 2 | Completed | Treatment | Endometriosis | 2 |
| 2 | Completed | Treatment | Prostate Cancer | 2 |
| 2 | Completed | Treatment | Uterine Leiomyomas | 1 |
| 3 | Active Not Recruiting | Treatment | Heavy Menstrual Bleeding / Uterine Leiomyomas | 2 |
| 3 | Active Not Recruiting | Treatment | Uterine Leiomyomas | 1 |
| 3 | Completed | Treatment | Uterine Leiomyomas | 1 |
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Endometriosis / Endometriosis related pain | 1 |
| 3 | Enrolling by Invitation | Treatment | Heavy Menstrual Bleeding / Uterine Leiomyomas | 1 |
| 3 | Enrolling by Invitation | Treatment | Uterine Leiomyomas | 1 |
| 3 | Recruiting | Treatment | Endometriosis related pain | 2 |
| 3 | Recruiting | Treatment | Prostate Cancer | 1 |
///////////Relugolix, TAK-385, JAPAN 2019, Relumina, レルゴリクス , PHASE 3
CONC(=O)NC1=CC=C(C=C1)C1=C(CN(C)C)C2=C(S1)N(CC1=C(F)C=CC=C1F)C(=O)N(C2=O)C1=CC=C(OC)N=N1