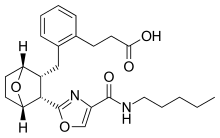
Ifetroban イフェトロバン
3-[2-({(1S,2R,3S)-3-[4-(Pentylcarbamoyl)-1,3-oxazol-2-yl]-7-oxabicyclo[2.2.1]hept-2-yl}methyl)phenyl]propanoic acid
- Molecular FormulaC25H32N2O5
- Average mass440.532 Da
- 143443-90-7;
Ifetroban is a potent and selective thromboxane receptor antagonist.[1]
Ifetroban has been used in trials studying the treatment of Skin Diseases, Autoimmune Diseases, Pathologic Processes, Scleroderma, Limited, and Scleroderma, Diffuse, among others.
This compound belongs to the class of organic compounds known as phenylpropanoic acids. These are compounds with a structure containing a benzene ring conjugated to a propanoic acid.
- OriginatorBristol-Myers Squibb
- DeveloperBristol-Myers Squibb; Cumberland Pharmaceuticals; Vanderbilt-Ingram Cancer Center
- ClassAntiasthmatics; Antihypertensives; Antiplatelets; Heterocyclic bicyclo compounds; Oxazoles; Small molecules
- Mechanism of ActionThromboxane A2 receptor antagonists
- Phase IIAsthma; Hepatorenal syndrome; Portal hypertension; Solid tumours; Systemic scleroderma
- DiscontinuedCoronary thrombosis; Peripheral vascular disorders; Thrombosis
- 12 Dec 2018Phase-II clinical trials in Solid tumours (Metastatic disease, Late-stage disease, Second-line therapy or greater, Recurrent) in USA (PO) (NCT03694249)
- 13 Nov 2018Efficacy and adverse events data from a phase II trial in Portal hypertension released by Cumberland Pharmaceuticals
- 03 Oct 2018Vanderbilt-Ingram Cancer Center and Cumberland Pharmaceuticals plans a phase II trial for Solid tumours (Metastatic disease, Late-stage disease, Second-line therapy or greater, Recurrent) (PO, capsule) (NCT03694249)
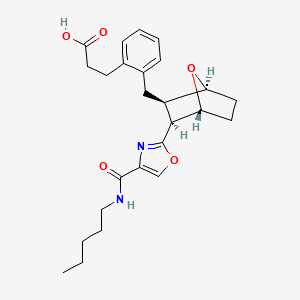
Ifetroban sodium
- Molecular FormulaC25H31N2NaO5
- Average mass462.514 Da
- Monoisotopic mass462.213074 Da

SYN
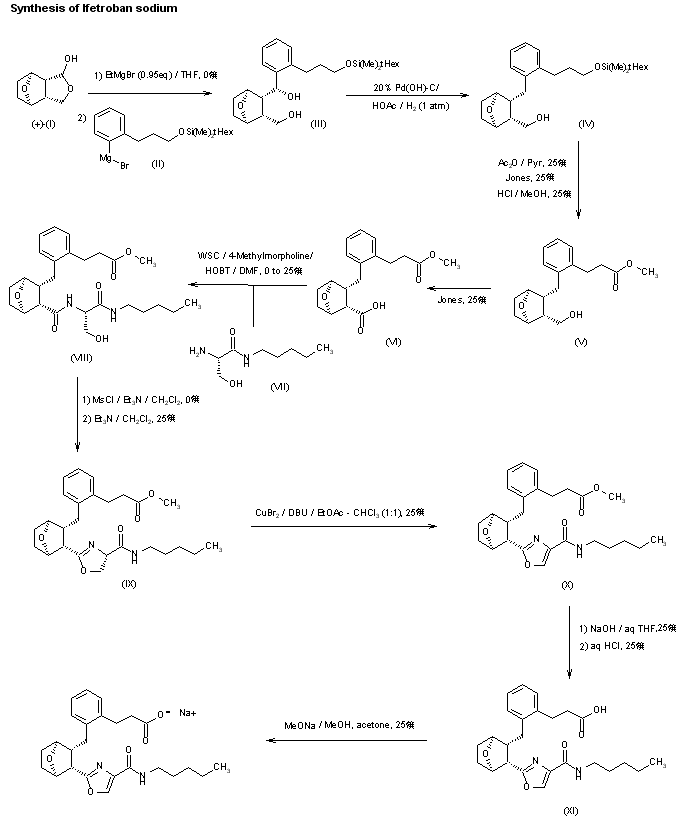
BMS-180291 sodium salt was prepared from optically active 7-oxabicyclo[2.2.1]heptane lactol (I): The interphenylene side chain was introduced by deprotonation of (I) with ethylmagnesium bromide (0.95 eq.) followed by treatment with excess aryl Grignard (II) to afford crystalline diol (III). The extraneous benzylic hydroxyl group in (III) was removed by reduction with hydrogen in the presence of Pearlman’s catalyst to give alcohol (IV). Transformation of the alpha-side chain silyloxy carbinol of (IV) to a carboxymethyl ester was accomplished by initial protection of the omega-side chain alcohol as the acetate (Ac2O/py) followed by oxidation under Jones conditions and then exposure of the resulting crude acetate-acid to methanolic hydrogen chloride to afford crystalline alcohol-ester (V). Oxidation of (V) under Jones conditions furnished acid-ester (VI). The oxazole side chain was introduced into (VI) via serine-derived amino alcohol (VII). Standard coupling of acid (VI) with (VII) mediated by water-soluble carbodiimide (EDAC) gave amide (VIII). Acyclic side chain intermediate (VIII) was converted into oxazole (X) in three steps by mesylation followed by treatment with triethylamine to furnish cyclized oxazoline (IX). Dehydrogenation of (IX) employing a novel oxidative protocol (1) involving treatment with a mixture of copper (II) bromide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in chloroform/ethyl acetate solvent yielded oxazole (X). Saponification of (X) followed by acidification afforded (BMS-180291) as a white solid which could be purified by recrystallization from acetonitrile. The water-soluble sodium salt (XI) was available as a precipitate from BMS-18091 by treatment with sodium methoxide/methanol in acetone.
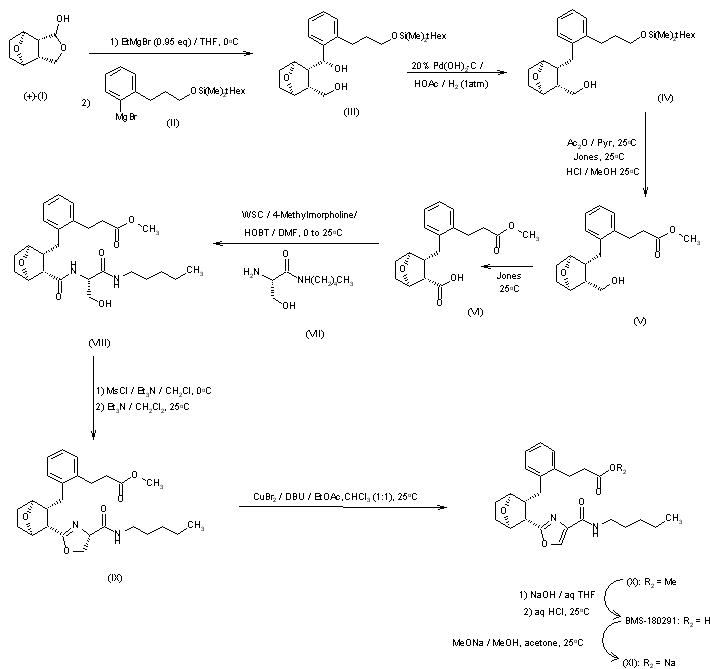
SYN
The interphenylene side chain was introduced by deprotonation of (I) with ethylmagnesium bromide (0.95 eq.) followed by treatment with excess aryl Grignard (II) to afford crystalline diol (III). The extraneous benzylic hydroxyl group in (III) was removed by reduction with hydrogen in the presence of Pearlman’s catalyst to give alcohol (IV). Transformation of the alpha-side chain silyloxy carbinol to a carboxy methyl ester was accomplished by initial protection of the omega-side chain alcohol as the acetate (Ac2O/pyr) followed by oxidation under Jones conditions and then exposure of the resulting crude acetate-acid to methanolic hydrogen chloride to afford crystalline alcohol-ester (V). Oxidation of (V) under Jones conditions furnished acid-ester (VI). The oxazole side chain was introduced into (VI) via serine-derived amino alcohol (VII). Standard coupling of acid (VI) with (VII) mediated by water-soluble carbodiimide (EDAC) gave amide (VIII). Acyclic side chain intermediate (VIII) was converted into oxazole (X) in three steps by mesylation followed by treatment with triethylamine to furnish cyclized oxazoline (IX). Dehydrogenation of (IX) employing a novel oxidative protocol involving treatment with a mixture of copper (II) bromide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in chloroform/ethyl acetate solvent yielded oxazole (X). Saponification of (X) followed by acidification afforded (XI) (BMS-180291) as a white solid which could be purified by recrystallization from acetonitrile. The water-soluble sodium salt was available as a precipitate from (XI) by treatment with sodium methoxide/methanol in acetone.
SYN
| Org Process Res Dev 1997,1(1),14 |
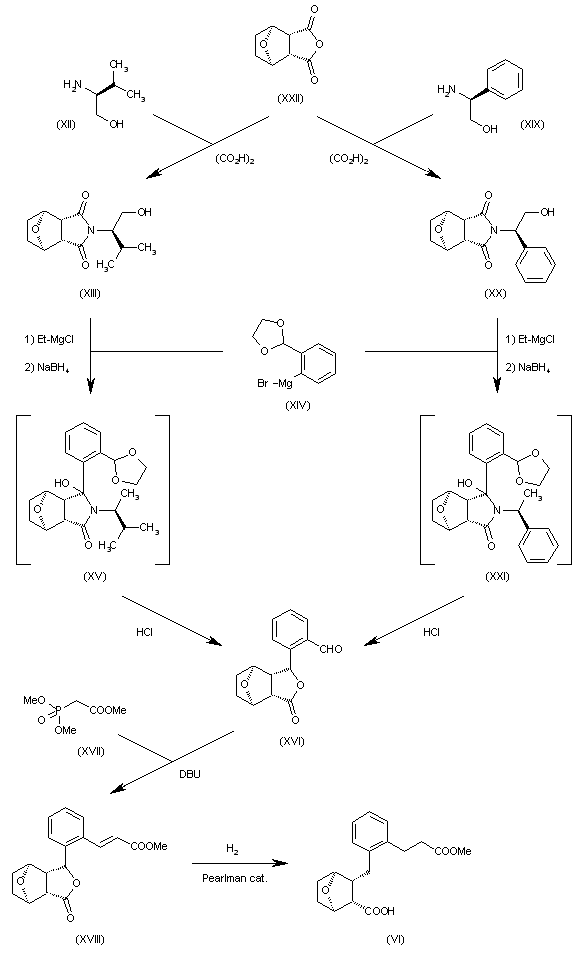
The synthesis of [1S-(1alpha,2alpha,3alpha,4alpha)]-2-[2-[2-(methoxycarbonyl)ethyl]benzyl]-7-oxabicyclo[2.2.1]heptane-3-carboxylic acid (VI), a key intermediate in the synthesis of 203961 [see scheme 20396101a] has been presented: This compound has been obtained by two similar ways: 1) The condensation of L-valinol (XII) with anhydride (XXII) catalyzed by oxalic acid gives imide (XIII), which is treated with ethylmagnesium chloride, the Grignard reagent (XIV) and NaBH4 yielding intermediate (XV). This intermediate, without isolation, is treated with HCl in THF to afford the substituted benzaldehyde (XVI), which is condensed with trimethyl phosphonoacetate (XVII) and DBU in acetonitrile giving the propenoic ester (XVIII). Finally, this compound is submitted to a simultaneous reduction and hydrogenolysis with H2 over a Pearlman catalyst in methanol to provide the target of [1S-(1alpha,2alpha,3alpha,4alpha)]-2-[2-[2-(methoxycarbonyl)ethyl]benzyl]-7-oxabicyclo[2.2.1]heptane-3-carboxylic acid (VI). 2) The preceding reaction sequence can also be performed using (S)-2-phenylglycinol (XIX) instead of the L-valinol (XII) yielding the previously reported benzaldehyde (XVI) through the imide (XX) and the nonisolated intermediate (XXI).
References
- ^ Dockens, RC; Santone, KS; Mitroka, JG; Morrison, RA; Jemal, M; Greene, DS; Barbhaiya, RH (August 2000). “Disposition of Radiolabeled Ifetroban in Rats, Dogs, Monkeys, and Humans”(PDF). Drug Metabolism and Disposition. 28 (8): 973–80. PMID 10901709. Retrieved 5 October 2016.
|
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C25H32N2O5 |
| Molar mass | 440.53 g/mol |
| 3D model (JSmol) | |
////////Ifetroban, BMS 18029, BMS 180291, BMS 180291A, BMS-180291-02, Boxaban, CPI 211, Hepatoren, Portaban, Vasculan, イフェトロバン
CCCCCNC(=O)C1=COC(=N1)C2C3CCC(C2CC4=CC=CC=C4CCC(=O)O)O3