
Selexipag
- Molecular FormulaC26H32N4O4S
- Average mass496.622 Da
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
セレキシパグ
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
FDA approves new orphan drug to treat pulmonary arterial hypertension
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]
In 2016, the EMA granted marketing authorization in the E.U. for this indication and launch took place shortly after in Germany and the United Kingdom. In Japan, Nippon Shinyaku received approval for the treatment of PAH in 2016.
Selexipag was approved by the U.S. Food and Drug Administration (FDA) on Dec 21, 2015, approved by European Medicine Agency (EMA) on May 12, 2016. It was originally developed by Nippon Shinyaku and then it was licensed to Actelion for co-development. It is marketed as Uptravi® by Actelion in US and EU.
Selexipag is a prostacyclin receptor (PGI2) agonist, which leads to vasodilation in the pulmonary circulation. It is indicated for the treatment of pulmonary arterial hypertension (PAH).
Uptravi® is available as tablets for oral use, containing 200, 400, 600, 800, 1000, 1200, 1400, or 1600 mcg of selexipag. The initial dose is 200 mcg twice daily, and increase the dose by 200 mcg twice daily at weekly intervals to the highest tolerated dose up to 1600 mcg twice daily.

ACT-333679 or MRE-269, the active metabolite of selexipag
SYNTHESIS DEPICT



PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
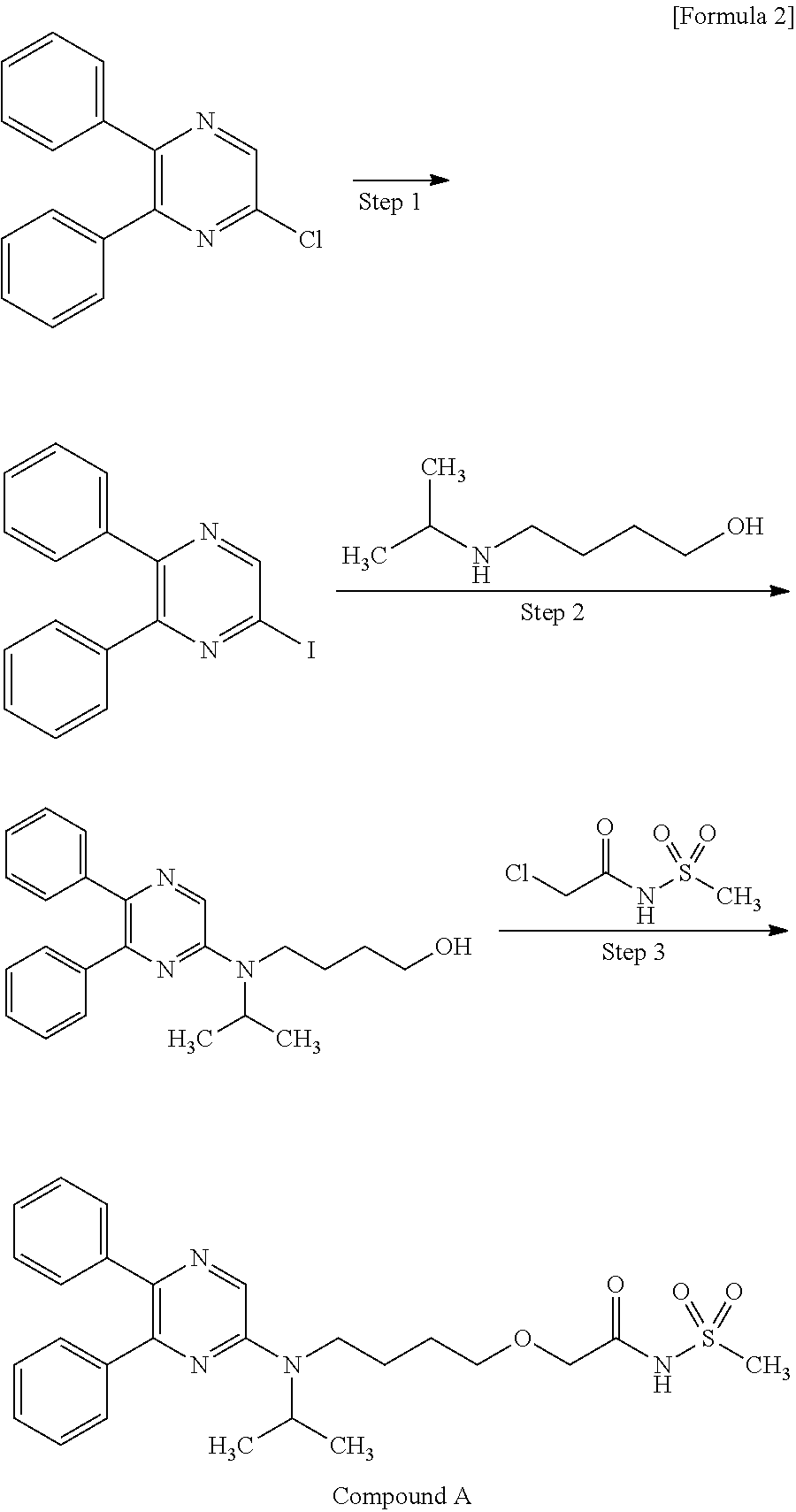
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a is the drug
2a is the drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
PATENT
Compound A Compound A can be produced , for example, by the method described in Patent Document 1, but can also be produced by the production method described below.
[
6-iodo-2,3-diphenylpyrazine can be produced by reacting 6-chloro-2,3-diphenylpyrazine with sodium iodide. This reaction is carried out in an organic solvent (for example, ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or a mixed solvent thereof) in the presence of an acid. As the acid to be used, for example, acetic acid, sulfuric acid, or a mixed acid thereof can be mentioned. The amount of sodium iodide used is, for example, suitably in the range of 1 mole to 10 moles, preferably in the range of 2 time moles to 3 times the amount of 1 mole of 6-chloro-2,3-diphenylpyrazine . The reaction temperature varies depending on the raw materials used and the type of acid, but is usually carried out within the range of 60 ° C. to 90 ° C. The reaction time varies depending on the starting materials used, the type of acid and the reaction temperature, but it is usually within the range of 9 hours to 15 hours.Step 2
5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] pyrazine can be prepared by reacting 6-iodo-2,3-diphenylpyrazine with 4-hydroxybutyl (isopropyl) amine. This reaction is carried out in an organic solvent (for example, sulfolane, N-methylpyrrolidone, N, N-dimethylimidazolidinone, dimethylsulfoxide or a mixed solvent thereof) in the presence of a base. Examples of the base used include sodium hydrogencarbonate, potassium hydrogen carbonate, potassium carbonate, sodium carbonate, and mixed bases thereof. The amount of 4-hydroxybutyl (isopropyl) amine to be used is, for example, suitably in the range of 1.5 mol to 5.0 mol per 1 mol of 6-iodo-2,3-diphenylpyrazine, It is within the range of 2 mol to 3 mol. The reaction temperature varies depending on the type of raw material and base used, but is usually carried out within the range of 170 ° C. to 200 ° C. The reaction time varies depending on the type of raw materials and base used and the reaction temperature, but it is usually within the range of 5 hours to 9 hours.Step 3
Compound A can be prepared by reacting 5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] pyrazine with N- (2-chloroacetyl) -methanesulfonamide. This reaction is carried out in an organic solvent (N-methylpyrrolidone, 2-methyl-2-propanol or a mixed solvent thereof) in the presence of a base. Examples of the base to be used include potassium t-butoxide, sodium t-butoxide or mixed bases thereof. The amount of N- (2-chloroacetyl) -methanesulfonamide used is, for example, 2 to 4 mol per 1 mol of 5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] It is suitable within the range, and preferably within the range of 2 mol to 3 mol. The reaction temperature varies depending on the type of raw material and base used, but is usually carried out within the range of -20 ° C. to 20 ° C. The reaction time varies depending on the kinds of raw materials and bases used and the reaction temperature, but it is usually within the range of 0.5 hour to 2 hours.Each compound used as a raw material in the above-mentioned production method of compound A is a known compound or can be produced according to a known method.
present invention can be obtained, for example, by the following method.
The salt of the present invention can be prepared by dissolving the compound A in an appropriate solvent (for example, an ether solvent (for example, dimethoxyethane, tetrahydrofuran), an ester solvent (for example, isopropyl acetate), an aromatic hydrocarbon (for example, toluene), acetonitrile After dissolving and adding a desired base, if necessary, the mixed solution is left to stand at room temperature or under cooling in the state of concentrating or stirring or leaving it stationary. The precipitate formed is collected by filtration , Followed by washing with an appropriate solvent to obtain the desired salt of the present invention. When cooling, not only cooling but also gradual cooling or rapid cooling may be effective in obtaining good crystals. It is also effective to obtain good crystals by adding an ether solvent (for example, t-butyl methyl ether), an ester solvent (for example, ethyl acetate), and an aromatic hydrocarbon (for example, toluene) There are cases.The amount of the solvent used for dissolving the compound A is suitably in the range of 10 ml to 300 ml with respect to the compound A 1 g, for example.
The amount of the base to be used for preparing the salt of the present invention is suitably in the range of 0.5 mol to 1.2 mol with respect to the mol of the compound A 1.
Further, the salt of the present invention, which is a crystal, can be obtained by, for example, the method described in Examples described later.
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.
Paper
J Med Chem 2015, 58(18): 7128
PATENT
WO 2018008042
https://patents.google.com/patent/WO2018008042A1/en
The present invention relates to an improved and novel processes for the preparation of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N-(methylsulfonyl) acetamide compound of formula- 1 , which is represented by the following structural formula- l .

Formula-
The present invention also relates to novel crystalline forms of the compound of formula- 1 and process for the preparation thereof.
Background of the Invention:
2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl) acetamide is known as Selexipag. It is developed by Nippon Shinyaku under the brand name of Uptravi®, for the treatment of pulmonary arterial hypertension.
2- {4-[(5,6-diphenylpyTazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl) acetamide was firstly described in US7205302B2 herein after referred as US ‘302. The said patent also describes its process for the preparation. According to this process the final product was obtained with low yield and purity.
US8791 122 (herein after referred as US’ 122) patent describes crystalline form-I, II and III of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N-(methylsulfonyl) acetamide. Because of drug compounds having, for example, improved stability, solubility, shelf life and in vivo pharmacology, are consistently sought, there is an ongoing need for new or pure salts, hydrates, solvates and polymorphic forms of existing drug molecules. The novel crystalline forms of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N- (methylsulfonyl) acetamide described herein help meet this requirement.
US ‘ 122 patent describes amorphous form of the compound of formula- 1 . This patent does not disclose any detailed process for amorphous form and PXRD pattern of amorphous compound of formula- 1 .

Examples:
Example-1 Preparation of 4-((5)6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol compound of formula-8
A mixture of 5-chloro-2,3-diphenylpyrazine (25 gm) compound of formula-7a and 4- (isopropyl amino)butan- 1 -ol (108 gm) was heated to 190-195°C and stirred the reaction i mixture for 10- 12 hours at same temperature. Cooled the reaction mixture to 25-35°C. To this reaction mixture n-heptane followed by water were added slowly at 25-30°C and stirred the reaction mixture for 2 hours at the same temperature. Filter the precipitated solid, washed with water and dried to get the title compound.
Yield: 30 gm.
Example-2: Preparation of tert-butyl 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino) butoxy)acetate
Potassium hydroxide solution (96.6 gm of potassium hydroxide dissolved in 175 ml of water) was added to the mixture of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan- l -ol (25 gm) and toluene ( 175 ml) at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Cooled the reaction mixture to 0-5°C. Tert-butyl bromoacetate (94 gm) was slowly added to the reaction mixture at 0-5°C and stirred the reaction for 60 minutes at same temperature. Raised the temperature of the reaction mixture to 25-30°C and maintained for 60 minutes. Both the aqueous and organic layers were separated. The aqueous layer was extracted with toluene and combined the organic layers. Organic layer was washed with hydrochloric acid solution followed by with aqueous sodium bicarbonate solution. Organic layer was dried with sodium sulphate and distilled off the solvent completely from the organic layer under reduced pressure to get the title compound.
Yield: 29 gm.
Example-3: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy) acetic acid compound of formula-6
Aqueous sodium hydroxide solution (7.5 gm of sodium hydroxide was dissolved in 80 ml of water) was added to the solution of tert-butyl 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl) amino)butoxy)acetate (30 gm) in methanol (290 ml) at 30-35°C. Heated the reaction mixture to reflux temperature and stirred for 3 hours at the same temperature. Distilled off solvent completely from the reaction mixture under reduced pressure and cooled the reaction mixture to 25-30°C. Water was added to the obtained compound and acidified the reaction mixture using diluted hydrochloric acid at the same temperature. Extracted the reaction mixture with ethyl acetate. The organic layer was washed with aqueous sodium chloride solution and dried with sodium sulphate. Distilled off the solvent from the organic layer under reduced pressure. Diisopropyl ether (60 ml) was added to the obtained compound at 25-30°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with diisopropyl ether and dried to get the title compound.
Yield: 19 gm.
Example-4: Preparation of 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropy.)amino]butoxy}- N-(methylsulfonyl) acetamide compound of formula-1
Triethylamine (9.6 gm) was added to the mixture of 2-(4-((5,6-diphenylpyrazin-2- yl)(isopropyl)amino)butoxy)acetic acid (10 gm), dichloro methane (100 ml), N,N- dicyclohexylcarbodiimide (4.9 gm), hydroxybenzotriazole (3.5 gm) and methane sulfonamide (3.39 gm) at 25-30°C and stirred the reaction mixture for 12 hours at the same temperature. Filtered the unwanted compounds from the reaction mixture and washed with dichloromethane. The organic layer was washed with water, followed by with aqueous citric acid solution and then washed with aqueous sodium chloride solution. Distilled off the solvent from the organic layer under reduced pressure. To this residue ethyl acetate (20 ml) and carbon (1 gm) were added at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with ethyl acetate. The obtained filtrate was slowly added to the mixture of n-heptane and water at 25-30°C and stirred for 10 hours. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 4.5 gm.
Example-5: Preparation of 2-{4-f(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}- N-(methylsulfonyl) acetamide compound of formula-1
Sodium t-butoxide (96.6 gm) was added to the mixture of n-methy pyrrolidinone (125 ml) and 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (25 gm) compound of formula-8 at 0-5°C and stirred the reaction for 20 minutes at the same temperature. 2-chloro- N-(methylsulfonyl)acetamide (23.7 gm) was slowly added to the reaction mixture at 0-5°C and raise the temperature of the reaction mixture to 25-30°C. Stirred the reaction mixture for 10-12 hours at 25-30°C and water was added to it at the same temperature. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with aqueous sodium chloride solution and distilled off the solvent from the organic layer under reduced pressure. To this residue ethyl acetate (50 ml) and carbon (2.5 gm) were added at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with ethyl acetate. The obtained filtrate was slowly added to the mixture of n-heptane and water at 25-30°C and stirred for 10 hours. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 14 gm.
Example-6: Preparation of 2-chIot*o- -(methylsulfonyl)acetamide
A mixture of methane sulfonamide (100 gm) and chloroacetyl chloride (356.4 gm) was heated to reflux temperature and stirred it for 10 hours at the same temperature. Cooled the reaction mixture to – 10 to -5°C and stirred it for 2 hours at the same temperature. Filtered the precipitated, solid, washed with toluene followed by n-heptane and dried to get the title compound.
Yield: 175 gm.
ExampIe-7: Purification of the compound of formula-1
Methanol (20 ml) was added to the compound of formula-1 (2 gm) at 25-30°C and heated to reflux temperature. Dichloromethane (3 ml) was added to the reaction mixture at reflux temperature and stirred for 15 minutes at the same temperature. Filtered the reaction mixture, distilled off the solvent from the filtrate under reduced pressure to get the title compound. Yield: 2 gm
Example-8: Preparation of N-isopropyI-5,6-diphenylpyrazin-2-amine (Formula-4) Isopropyl bromide (5.5 gm) was added to the mixture of 2-amino -5,6-diphenylpyrazine ( 10 gm), potassium tert-butoxide (9 gm) and dimethylformamide (50 ml) at 25-30°C, slowly heated to 80-85°C and stirred the reaction mixture for 6 hours at same temperature. The reaction mixture was cooled to 10- 15°C, diluted the reaction mixture with water and stirred it for 2 hours at the same temperature. Filtered the obtained solid and dried to get the title compound.
Yield: 9.5 gm
ExampIe-9: Preparation of N-isopropyl-5,6-diphenylpyrazin-2-amine (Formula-4)
A mixture of 5-chloro-2,3-diphenylpyrazine ( 10 gm), isopropyl amine (7.5 gm) and potassium carbonate (10.5 gm) and dioxane (50 ml) were heated to 40-45°C and stirred the reaction mixture for 12 hrs at the same temperature. The reaction mixture was cooled to 10- 15°C, diluted with water and extracted with dichloromethane. Combined the organic layers was washed with aqueous sodium hydrochloride solution and dried over anhydrous sodium sulphate. Distilled off the solvent completely from the organic layer under reduced pressure to provide the title compound.
Yield: 9 gm
Example-10: Preparation of 2-(4-chlorobutoxy)aceticacid (Formula-5a)
2-bromoaceticacid (10 gm) was slowly added to a mixture of l-chlorobutan-4-ol (7.2 gm), potassium carbonate (26.5 gm) and acetonitrile (50 ml) at 25-30°C. The reaction mixture was heated to 75-80°C and stirred the reaction mixture for 6 hours at same temperature. The reaction mixture was cooled to 25-30°C and diluted with , water. Acidified the reaction mixture using diluted hydrochloric acid at 25-30°C. The reaction mixture extracted with dichloromethane. Combined the organic layers was dried over anhydrous sodium sulphate and distilled off the solvent under reduced pressure to provide the title compound.
Yield: 10.5 gm.
Example-11: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino) butoxy)acetic acid (formula-6)
A mixture of N-isopropyl-5,6-diphenylpyrazin-2-amine (8 gm), potassium carbonate (7.5 gm) and acetonitrile (40 ml) was stirred for 1 hr at 25-30°C. A solution of 2-(4-chlorobutoxy) aceticacid (5.4 gm) in acetonitrile (15 ml) was slowly added to the reaction mixture at 25- 30°C. Heated the reaction mixture to reflux and stirred for 12 hours at the same temperature. The reaction mixture was cooled to 10-15°C and diluted with wateT. Acidified the reaction mixture using diluted hydrochloric acid and extracted the reaction mixture using ethyl acetate. Combined the organic layers and dried over sodium sulphate. Distilled off the solvent completely from the organic layer to get the title compound.
Yield: 8.5 gm
Example-12: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyt)amino)butoxy)- N-(methylsulfonyl)acetamide (formula-1)
A mixture of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy)acetic acid (5 gm), HATU (5.4 gm), triethylamine (1.5 gm) and dimethylformamide (20 ml) was stirred for 1 hr at 5-10°C under nitrogen atmosphere. Methane sulfonamide (5.2 gm) was slowly added to the reaction mixture at 5-10°C and stirred for 12 hrs at the same temperature. The reaction mixture was diluted with water and stirred for 2 hrs. The precipitated solid was filtered and dried to get the title compound.
Yield: 4.5 gm
Example-13: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl) (isopropyl) amino) butoxy) acetonitrile (Formula-12)
To the mixture of 4-((5,6-diphenylpyrazin-2-yI)(isopropyl)amino)butan-l-ol ( 10 gm), tetrabutyl ammoniumbromide (0.2 gm), potassium carbonate (7.6 gm) and acetone (50 mL), chloroacetonitrile (3.2 gm) was added at 25-30°C. Heated the reaction mixture to reflux temperature and stirred the reaction mixture for 6 hrs at the same temperature. The reaction mixture was cooled to 10- 15°C and filtered the reaction mixture. Distilled off the solvent completely from the filtrate to get the tile compound.
Yield: 9 gm
Example-14: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy) acetic acid (formula-6)
Sodium hydroxide (3.5 gm) was added to a solution of 2-(4-((5,6-diphenylpyrazin-2-yl) (isopropyl) amino) butoxy) acetonitrile (8 gm) in methanol (60 ml) and water (30 ml). The reaction mixture was heated to 65-70°C and maintained for 6 hrs. The reaction mixture was cooled to 10°C, acidified with diluted hydrochloric acid and stirred at same temperature for 2 hr. The obtained solid was filtered and dried to provide the title compound.
Yield: 7.5 gm
Example-15: Preparation of 2-chloro-N-(methylsulfonyl)acetamide (Formula-16)
The mixture of methane sulfonamide (50 gm) and chloroacetyl chloride (92 gm) was heated to 1 10-1 15°C and stirred the reaction mixture for 7 hours at the same temperature. The reaction mixture was cooled to 25-30°C and dichloromethane was added to the reaction mixture at the same temperature. Cooled the reaction mixture to 15-20°C and stirred for 1 hour at the same temperature. Filtered the precipitated solid and washed with dichloromethane. The obtained solid was recrystallized using dichloromethane to get pure title compound. Yield: 80 gm. M.R.: U0- 1 15°C. Purity by HPLC: 98.85%.
Example-16: Preparation of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (Formula-8)
The mixture of 5-chloro-2,3-diphenylpyrazine ( 100 gm) and 4-(isopropylamino)butan-l -ol (245.5 gm) was heated to 190-195°G and stirred the reaction mixture for 12 hours at the same temperature. The reaction was cooled to 25-30°C and n-heptane was added to the reaction mixture. The reaction mixture was further cooled to 10-15°C, water was slowly added to the reaction mixture and stirred for 2 hours at the same temperature. Filtered the precipitated solid and washed with water. Dichloromethane (300 ml) was added to the obtained solid and stirred for 5 minutes. Both the organic and aqueous layers were separated. The organic layer was dried with sodium sulphate, distilled off the solvent from the organic layer completely under reduced pressure and co-distilled with n-heptane. 400 ml of n-heptane was added to the obtained compound at 25-30°C, heated the reaction mixture to 45-50°C and stirred for 30 minutes at the same temperature. The reaction mixture was cooled to 15-20°C and stirred for 2 hours at the same temperature. Filtered the solid, washed with n-heptane and dried to get the title compound.
Yield: 82 gm. M.R.: 100-105°C. Purity by HPLC: 95.4%.
Example-17: Purification of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (Formula-8)
n-Heptane (750 ml) was slowly added to pre-cooled solution of 4-((5,6-diphenylpyrazin-2- yl)(isopropyl)amino)butan- l -ol (100 gm) in acetone (250 ml) was cooled to 0-5°C. Stirred the reaction mixture for 4 hours at the same tempereature. Filtered the precipitated solid, washed with n-heptane and dried to get the pure title compound.
Yield: 54 gm. Purity by HPLC: 99.92%.
Example-18: Preparation of crystalline form-L of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of pre-cooled n-heptane at 0- 5°C. Stirred the reaction mixture for 6 hr at 0-5°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 9 gm; PXRD of the obtained compound is depicted in figure- 10 and DSC thermogram is depicted in figure- 1 1. Example-19: Preparation of crystalline form-P of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of pre-cooled n-heptane at 0- 5°C. Stirred the reaction mixture for 36 hours at 0-5°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 9 gm; PXRD of the obtained compound is depicted in figure-7, its IR is depicted in figure-8 and its DSC is depicted in figure-9.
Example-20: Preparation of crystalline form-P of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of n-heptane at 30-40°C.
Stirred the reaction mixture for 36 hours at 30-40°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 9 gm; PXRD of the obtained compound is similar to the figure-7.
Example-21 : Preparation of amorphous form of compound of formula-1
Melting the compound of formula-1 ( 10 gm) at 140- 145°C under reduced pressure for 15 minutes and the above obtained oily residue was cooled to 0-5°C. Unload the obtained compound and dried to get the title compound. Yield: 9 gm; Purity by HPLC: 99.74%. PXRD of the obtained compound is depicted in figure-5 and IR is depicted in figure-6.
Exaniple-22: Preparation of crystalline form-I of compound of formula-1
Melting the compound of formula-1 (5 gm) at 140-145°C under reduced pressure for 15 minutes. 50 ml of n-heptane was added to the above obtained oily residue at 115-120°C.
Stirred the reaction mixture for 20 minutes at 1 15- 120°C. Cooled the reaction mixture to 25-
30°C and stirred for 60 minutes at the same temperature. Further cooled the reaction mixture to 0-5°C and stirred the reaction mixture for 60 minutes at the same temperature. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 4 gm; Purity by HPLC: 99.68%.
PATENT
CN 108675964
PATENT
CN 106316967
PATENT
WO 2017029594
PATENT
Form-I II III
https://patents.google.com/patent/US8791122B2/en
PATENT
https://patents.google.com/patent/WO2018022704A1/en
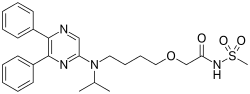
Selexipag has the chemical name 2-{4-[(5,6-diphenylpyrazin-2- yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide. Selexipag has the following chemical structure:

[0004] Selexipag is being developed by Actelion and Nippon Shinyaku for the treatment of arteriosclerosis obliterans, pulmonary hypertension and Raynaud’s disease secondary to systemic sclerosis.
[0005] Selexipag is disclosed in US 7,205,302. US 8,791,122, US 9,284,280 and US 2014- 0155414 disclose polymorphs of Selexipag, denominated forms I, II and III. WO
2017/040872 discloses form IV and V of Selexipag.
xample 1: Preparation of Selexipag
[00126] A. Route 1
[00127] Crude Selexipag can be obtained by any method known in the art, for example by the method described in US 7,205,302 or according to the following.
[00128] B. Route 2
[00129] Step a: Preparation of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl)amino)butan-l-ol
[00130] To 50 g (0.161 mol) of 5-bromo-2,3-diphenylpyrazine, 116 g (0.884 mol, 5.5 eq/mol) of 4-(isopropylamino)-butan-l-ol and 13.33 g of KI (0.080 mol, 0.5 Eq/mol) were added. The reaction mixture was stirred, warmed and then heated up to 140°C for about 18- 20 hrs. The reaction was monitored by TLC up to completion (starting material about 1% by TLC). The reaction mixture was cooled down to room temperature. After the reaction was completed, the following work up step was performed:
[00131] Option 1 : Ethyl acetate was added (500 mL, 10 vol) and the organic phase was washed with water (150 mL, 3 vol). The organic phase was separated and aqueous phase was extracted with ethyl acetate (150 mL, 3 vol). The organic phases were joined and washed with water (200 mL, 2 vol) three times.
[00132] The solvent was distilled off under vacuum at not more than (“NMT”) 40°C until 1 vol (oil appearance).
[00133] Option 2: The material (mixture) was dissolved in acetone (250 mL, 5 vol), the solution obtained was cooled down to 0°C to 5°C and anti-solvent / water was added (1000 mL, 20 vol) for 40 minutes, then the suspension was stirred for about 30 minutes at about 0°C-5°C. The solid material was filtered and washed with water (200 mL, 4 vol). Crude wet product was obtained as yellow solid yielding 101.8 % WY (87 % MY), HPLC purity 90.8% on area at this stage.
[00134] The crude material, obtained in either of the above described options, was purified through crystallization from acetone :«-heptane as follows: to a solution of 4-((5,6-diphenyl- pyrazin-2-yl)(isopropyl)amino)butan-l-ol crude in acetone (175 mL, 3.5 vol) at 0°C – 5°C, hexane (600 mL, 12 vol) dropwise in about 120 min was added, then the precipitated mixture was cooled down to about -10°C and stirred for about 60 min. The product was filtered off and washed with hexane (250 mL, 5 vol) and dried under vacuum at 25°C. Pure product was obtained as yellowish solid yielding overall 77.2%, (66.5% MY), HPLC purity 98.2% on area.
[00135] Step b: Preparation (2-bromo-N-(methylsulfonyl)-acetamide)
[00136] To a suspension of 50 g (0.526 mol) of methanesulfonamide in toluene (625 mL, 12.5 vol) and isopropyl acetate (625 mL, 12.5 vol), 159.1 g (0.789 mol) of bromo-acetyl- bromide (“BAB”) was added under nitrogen atmosphere. The reaction mixture was heated up to about 90°C for about 8 hours under a nitrogen stream. The reaction was monitored by TLC up to completion (starting material about 1% by TLC). The reaction mixture was cooled down to about 40°C and concentrated under vacuum until 10 volumes. Subsequently, toluene was added (250 mL, 5 vol) and distilling off solvents is carried out at NMT 30°C until 10 volumes. Then was added dichloromethane (100 mL, 2 vol) and the mixture was cooled down at 0°C and is stirred for 90 min. The solid was filtered and washed with
dichloromethane (100 mL, 2 vol). Crude product was obtained as beige solid material yielding 187% WY (83% MY), HPLC purity 99.2% at this stage. [00137] The crude material (83 g) was purified through re-slurring with dichloromethane (166 mL, 2 vol; preferably 332 mL, 4 vol) by stirring at about 32°C for about 60 min. The crystallization mixture was cooled down to about 0°C-5°C and stirring for 30 min, filtered off and washed with dichloromethane (100 mL, 2 vol). Subsequently, the material was dried at 35°C for 24 hours. Pure and dried material was obtained as white off solid yielding overall 173%, (77% MY), HPLC purity 99.6 % on area.
[00138] Step c: Preparation of (2-[4-[(5,6-diphenyl-2-pyrazinyl)(l- methylethyl)amino]butoxy]-N-(methylsulfonyl)-acetamide) – Selexipag
[00139] To 10 g (0.028 mol) of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl)amino) butan-l-ol was added a strong base (6.0 eq/mol), previously suspended in an appropriate solvent, within a range of from -10°C to 40°C under a nitrogen atmosphere and stirred for 60 min. Then, a solution of 17.9 g (3.0 eq/mol) of 2-bromo-N-(methylsulfonyl)-acetamide, previously dissolved in the same solvent, is added dropwise within a range of from 120 tol 80 min, controlling the exothermic temperature. The reaction was monitored by TLC up to completion. Subsequently, the mixture reaction was cooled down around 5°C and water is added by controlling the exotherm (NMT 15°C). Finally, an acetic acid solution was added and the suspension was stirred for about 60 min at 0°C -5°C. The product (crude) was filtered off and washed with water. An amorphous solid was obtained. The crude product was purified by crystallization from ethanol:THF.
[00140] Step d: Purification of Selexipag
[00141] Crude Selexipag can be purified by crystallization in an organic solvent for example alcohols such as ethanol, iso-amyl alcohol, iso-propyl alcohol, butanol; ethers such as tetrahydrofuran, hydrocarbons such as heptane and mixed solvents thereof.
[00142] C. Route 3
[00143] 33.3 g (0.297 mol, 6.0 eq/mol) of potassium tert-butoxide were dissolved in DMF (2.8 vol) in a flask (500 mL) under nitrogen atmosphere and stirred for 15 min. Then, a solution of 17.9 g (0.049 mol, 1.0 eq/mol) of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl) amino) butan-l-ol (SLX-4) dissolved in DMF (1.2 vol) was added in one portion. The reaction mixture was stirred for 60 min within a temperature range from 20°C to 25°C at 150 rpm Then, a solution of 32.1 g (0.15 mol, 3.0 Eq/mol) of 2-bromo-N-(methylsulfonyl)- acetamide (SLX-9), previously dissolved in DMF (1.3 vol), was added dropwise for 120 minutes by controlling the temperature (exothermic process).
[00144] The reaction mixture was quenched with cool water (0.33 vol), transferred into a flask of more capacity (1000 mL) and placed in an ice bath. Cool water (38.32 vol) was added to the reaction mixture and the pH was adjusted to 5.0 with AcOH (0.33 vol). The mixture was stirred at 300 rpm for 40 min. Then, the flask with the reaction mixture was stored in the refrigerator at 8°C. After 8h, the solid was filtered and washed with cool water (5 vol, 2 times). The crude product (yellow solid) was drained (i.e. dried) for 30 min and was stored at 8°C.
Example 2: Preparation of crystalline Selexipag Form IV
[00145] A. Route 1
[00146] 3.0 g of Selexipag was dissolved in dimethylformamide (“DMF”) (12 mL, 4 vol). The obtained solution was added dropwise to a pre-cooled acetic acid solution (0.06 M, 120 mL, from 2°C to 8°C) to obtain a suspension. The suspension was stirred within a range of from 2°C to 8°C for 30 min; then the material was filtered, washed with water (10 mL, 3.3 vol) and drained (i.e. dried) for 10 minutes. The solid material (amorphous) was suspended in heptane (25 mL, 7.5 vol) and the obtained suspension was stirred for 30 minutes at room temperature. The material was filtered, washed with heptane (20 mL, 6.6 vol) and drained (i.e. dried) under vacuum for at least 30 minutes at room temperature to obtain the Form IV Crystal.
[00147] B. Route 2
[00148] Crude Selexipag (1.0 g, amorphous solid, obtained from the synthesis) was dissolved in ethyl acetate (5 vol, 5 mL), then water was added (10 vol, 10 mL) into the solution, the mixture was stirred for about 10 minutes and the pH was adjusted to a range of from 8.0 to 9.0 by titration with K2CO3 solution. The phases were separated; the pH of the aqueous phase was adjusted to a range of from 3.5 to 5.0 by titration with acetic acid. Then, ethyl acetate (10 vol, 10 mL) was added into the aqueous phase, the obtained mixture was stirred and the phases were separated. The organic phase was distilled off under reduced pressure (from 2 to 3 volumes), and a solution was obtained. The obtained concentrated solution was quickly added to a mixture (suspension) of Form IV in ^-heptane (17 mL, 17 vol), over a period of less than 5 minutes, (the suspension temperature was of from 15°C to 25°C), and a suspension was obtained. The obtained suspension was stirred (155rpm) for 90 minutes at a temperature of from 0°C to 5°C. The suspension was filtered, washed with heptane, squeezed for 15 minutes and dried at 25°C, under vacuum, for about 14 hours. The product was analyzed by PXRD – Form IV was obtained.
[00149] The above procedure can be performed by dissolving the crude amorphous starting material in any suitable organic solvent, for example ester solvent. Example 3: Preparation of (2-[4-[(5,6-diphenyl-2-pyrazinyl)(l- methylethyl)amino] butoxy] -N-(methylsulfonyl)-acetamide) – Selexipag

SLX-4 SLX-9 SLX-6
[00150] 9.2 grams (0.082 mol, 5.9 eq/mol) of potassium tert-butoxide were combined with DMF (2.7 vol, 13.5 mL) in a flask (50 mL) under nitrogen atmosphere and a suspension was formed and was stirred for 20 min. Then, 5.0 g (0.014 mol, 1.0 eq/mol) of 4-((5,6-diphenyl- pyrazin-2-yl)(isopropyl)amino)butan-l-ol (SLX-4) as solid powder was added under nitrogen atmosphere. The reaction mixture was stirred for 60 min within a temperature range from 20°C to 25°C and at 170 rpm. Then, a solution of 8.9 g (0.041 mol, 3.0 eq/mol) of 2-bromo- N-(methylsulfonyl)-acetamide (SLX-9), previously dissolved in DMF (1.3 vol, 6.5 mL), was added dropwise for 120 minutes by controlling the temperature (exothermic process). After the end of addition, the reaction was completed, and the reaction mixture was quenched with cold water (0.5 vol, 2.5 mL), subsequently transferred into a flask of more capacity (500 mL) and placed into an ice bath. Cold water (40 vol, 200 mL) was added into the suspension and the pH was adjusted within the range from 4.0 to 5.0 with acetic acid. The obtained mixture was stirred for 120 min. The crude amorphous product was collected by filtration and washed twice with cold water (5 vol, 25 mL). The product was drained (i.e. dried) for 30 min and isolated as a yellow-brown solid which was stored within the range from 2°C to 8°C for approximately 17 hours. Then, the crude amorphous material was dissolved in ethyl acetate (15 vol, 75 mL) and water was added into the solution (30 vol, 150 mL). The pH was adjusted from 8.0 to 9.0 by addition of potassium carbonate solution, the phases were separated and the aqueous phase was washed twice with ethyl acetate (7.5 vol, 37.5 mL). The pH of the final aqueous phase was adjusted to a range from 4.0 to 5.0 with acetic acid. Then, ethyl acetate was added (30 vol, 150 mL) and the phases were separated. The organic phase was washed twice with water (7.5 vol, 37.5 mL). The organic phase was distilled off under reduced pressure (from 6 to 7 volumes, or from 6 to 15 volumes) and a solution was obtained.
[00151] In a different flask (capacity of 250 mL with a PTFE stirrer blade), a suspension of 0.05 g of Selexipag Form IV in ^-heptane (30 volumes, 150 mL) was stirred for 60 minutes within the range 0°C to 5°C and this suspension was added into the above ethyl acetate concentrated solution at room temperature over a period of less than 5 minutes. The final suspension was cooled down to 0°C to 5°C and stirred (220 rpm) for 120 minutes. The solid product was filtered off and washed twice with cold heptane (5 vol, 25 mL). The product was drained (i.e. dried) overnight. The product was analyzed by PXRD – Form VI was obtained, PXRD pattern is depicted in Figure 1.
PATENT
CN105949135
https://patents.google.com/patent/CN105949135A/en

Example 1
[0027] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0028] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] butanol -1_ (20 (^, 0.09111 〇1) and tert-butyl bromoacetate (21 · lg, 0 llmol) solution. The reaction was stirred for 2 hours to burn dichloromethane (90mL), was added tetrabutylammonium chloride (0.72g, 2.6mmol), potassium hydroxide (7.3g, 0.13mol) and water (12.0g), the reaction mixture was 25 ° C The reaction solution was concentrated under reduced pressure and rotary evaporated to dryness and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl yl) aminobutoxy] acetate, as a pale yellow oil (26.6 g of), a yield of 89.0%, the reaction formula of this step is as follows:
[0029]

[0030] B) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0031] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (26. (^, 0.075! 11〇1) was dissolved in methanol (50 mL), was added sodium hydroxide solution (NaOH = 3 · 3g, 0 · 08mol; water 9 · 0g), was heated to 80 ° C for 6 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid (20.7 g of), a yield of 95.0%, the reaction formula of this step is as follows:

[0033] C) Preparation of 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0034] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (20 (^, 0.07 11〇1) and a hoot “-.! P sitting carbonyldiimidazole (14.0g, 0.09mo 1 ) was dissolved in tetrahydro-thiopyran Misaki (70 mL), with stirring, was added methyl sulfonamide (7.9g, 0.08mol), the reaction mixture was 90 ° C the reaction stirred for 18 hours, the reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (21.2 g), yield 83.7%, the reaction formula of this step is as follows:

[0036] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (20 (^, 0.055111〇1. ) and dissolved in methanol (1101 ^), trifluoroacetic acid (6.88,0.06111〇1), 65 ° (: the reaction stirred for 6 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (200 mL), cooled to 0 ° C crystallization for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in methanol (40 mL), was added 5 – chloro-2,3-diphenyl-pyrazine (16 · 0g, 0 · 06mol), N, N- diisopropylethylamine (15 · 5g, 0 · 12mol), the reaction mixture was stirred reactor 8 100 ° C hours, the reaction was cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (25.0 g of), a yield of 92.3%, the reaction step formula as follows:
[0037]

[0038] Example 2
[0039] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0040] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (23 (^, 0.10111 〇1) and tert-butyl bromoacetate (25 · 2g, 0 · 13mol) was dissolved. burning in 1,2-dichloroethane (110mL), was added tetrabutylammonium bromide (1 · lg, 3 · 5mmol), sodium hydroxide (6.4g, 0.16mol) and water (14.0g), the reaction mixture was 30 ° C The reaction was stirred for 3 hours, the reaction solution was concentrated by rotary evaporation to dryness under reduced pressure and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxy butoxycarbonyl) (isopropyl) aminobutoxy] acetate, as a pale yellow oil (30.3 g of), a yield of 88.2%, the reaction of the present step is the same formula as in Example 1;
[0041] B) Preparation of [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0042] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (30. (^, 0.09! 11〇1) was dissolved in ethanol (85 mL), was added potassium hydroxide solution ( 1 (! = 5.78,0.10111〇1 01; 128 water), heated to 75 ° (: 7 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid, an off-white solid (23.5 g of), a yield of 93.7%, the reaction of the present step is the same formula as in Example 1;
[0043] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0044] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (23 (^, 0.08 11〇1) and Chi ^ -! Dicyclohexyl carbodiimide (22. lg, 0. llmol) was dissolved in chloroform (120 mL), with stirring, was added methyl sulfonamide (9.8g, 0. lOmol), the reaction mixture was 80 ° C the reaction stirred for 19 hours, the reaction solution was concentrated by rotary evaporation to dryness, ethyl acetate was added and extracted dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide, off-white the solid (24.8 g of), a yield of 85.0%, the reaction of the present step is the same formula as in Example 1;
[0045] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (24 (^, 0.065111〇1. ) and dissolved in ethanol (1601 ^), trifluoroacetic acid (9 (^, 0.08111〇1.), 70 ° (: the reaction was stirred for 7 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (260 mL of), cooled crystallization to 0 ° C for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in ethanol (90 mL) , 5-chloro-2,3-diphenyl-pyrazine (! 11〇1 21.8 8,0.08), triethylamine (14.98,0.15111〇1), the reaction mixture was 100 ° (: The reaction was stirred for 18 hours, the reaction solution cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (29.6 g of), a yield of 91.0%, the reaction of the present step is the same formula as in Example 1 .
[0046] Example 3
[0047] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0048] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (12g, 0.05mol) and t-butyl bromoacetate (12.1g, 0.06mol) was dissolved in chloroform (70mL), was added tetrabutylammonium iodide (0 · 5g, 1 · 3mmol), lithium hydroxide (1 · 7g, 0 · 07mol) and water (6.5 g of), the reaction mixture was stirred 20 ° C for 4 hours, the reaction solution under reduced pressure concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl) aminobutyrate oxygen yl] acetate, as a pale yellow oil (15.6 g of), a yield of 86.8%, the reaction of the present step is the same formula as in Example 1;
[0049] B) Preparation of [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0050] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (15.0g, 0.04mol) was dissolved in isopropanol (40mL), was added a solution of lithium hydroxide (LiOH = 1 · 3g, 0 · 05mol; water 6 · 0g), was heated to 70 ° C for 8 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl) aminobutyrate oxy] acetic acid as an off-white solid (11.7 g), 93.0% yield, this step is the same reaction scheme of Example 1;
[0051] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0052] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (11 (^, 0.04! 11〇1) and 1- (3-dimethylaminopropyl) -3- ethylcarbodiimide (8.38,0.05111〇1) was dissolved in acetonitrile (4〇1111 ^), with stirring, was added methyl sulfonamide (5.] ^, 0.05mol), the reaction mixture was 95 ° C the reaction stirred for 22 hours, The reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (11.7 g), yield 84.2%, the reaction of the present step is the same formula as in Example 1;
[0053] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (11 (^, 0.03111〇1. ) and dissolved in dichloromethane (601 ^), trifluoroacetic acid (4.48,0.04111〇1), 50 ° (: the reaction was stirred for 10 hours to water (120 mL completion of the reaction, the reaction liquid was added to a stirred), cooled to crystallization 0 ° C for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in tert-butanol (40 mL ), 5-chloro-2,3-diphenyl-pyrazine (9.68,0.036 11〇1), 4-dimethylaminopyridine (8.18,0.07111〇1), the reaction mixture was 110 ° (:! reaction was stirred for 14 hours the reaction was cooled to room temperature, water (15 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (13.5 g of), a yield of 90.5%, the reaction in this step is the same formula Example 1.
[0054] Example 4
[0055] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0056] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (15 (^, 0.065111〇1) and t-butyl bromoacetate (17.78,0.09111〇1) was dissolved in toluene (701] 11 ^), was added tetrabutylammonium hydrogen sulfate (0.888,2.61] 11] 1〇1), potassium carbonate (15.2 area, 0.1 lmol) and water (9.5 g of), the reaction mixture was stirred 40 ° C for 1.5 hours, the reaction solution was concentrated under reduced pressure and rotary evaporated to dryness and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl propyl) aminobutoxy] acetate, as a pale yellow oil (19.6 g of), in the same reaction formula in this step a yield of 87.5% in Example 1;
[0057] B) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0058] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (19 (^, 0.055! 11〇1) was dissolved in tert-butanol (60 mL), hydroxide solution of cesium (CsOH = 11. lg, 0.07mol; water, 8.0 g), the reaction was heated to 75 ° C for 6.5 hours cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid, an off-white solid (15.0 g of), a yield of 94.2%, the reaction of the present step is the same formula as in Example 1;
[0059] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0060] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (15.0g, 0.05mol) and diazabicyclo 1,8_
[5.4.0] – | -7- dilute (9.5 region, 0.06111〇1) was dissolved in toluene (8〇1111 ^), with stirring, was added methyl sulfonamide (5.7 region, 0.06mo 1), the reaction mixture was 105 ° C for 16 hours, the reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (16.6 g of), 87.3% yield, this step is the same reaction scheme of Example 1;
[0061] D) Preparation of SIPA Seiler: 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (16 (^, 0.04111〇. 1) and dissolved in ethyl acetate (^ 1,301,111), trifluoroacetic acid (5.78,0.051] 1〇1), 80 <€ the reaction was stirred for 5 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (150 mL), was cooled to 0 ° C crystallization for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), then dissolved in isopropanol (50 mL), was added 5-chloro-2,3-diphenyl-pyrazine (13.58,0.05 11〇1!), a hoot dimethylaniline (12.28,0.10111〇1), the reaction mixture was 95 ° (: The reaction was stirred 12 hours, the reaction was cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (19.7 g of), a yield of 91.0%, the reaction step formula in Example 1.
//////////////
Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
-
Publication numberPriority datePublication dateAssigneeTitleWO2016193994A12015-05-292016-12-08Megafine Pharma (P) Ltd.Amorphous selexipag and process for preparation thereofWO2017040872A12015-09-032017-03-09Teva Pharmaceuticals International GmbhSolid state forms of selexipagWO2017042731A12015-09-102017-03-16Lupin LimitedAmorphous form of selexipag and solid dispersion thereofWO2018008042A1 *2016-07-052018-01-11Maithri Drugs Private LimitedNovel process for the preparation of 2-{4-[(5,6-diphenyl pyrazin-2-yl)(isopropyl)amino]butoxy}-n-(methylsulfonyl)acetamide and novel polymorphs thereofWO2018015974A12016-07-202018-01-25Mylan Laboratories LimitedPolymorphic forms and amorphous solid dispersion of selexipagWO2018022704A12016-07-262018-02-01Teva Pharmaceuticals International GmbhCrystalline form vi of selexipagWO2018078383A12016-10-272018-05-03Cipla LimitedPharmaceutical composition comprising amorphous selexipagFamily To Family CitationsWO2017029594A1 *2015-08-172017-02-23Dr. Reddy’s Laboratories LimitedProcesses for preparation of selexipag and its amorphous formWO2017042828A3 *2015-09-102017-04-27Megafine Pharma (P) Ltd.Process for the preparation of selexipagWO2017168401A1 *2016-04-012017-10-05Honour (R&D)Process for the preparation of diphenylpyrazine derivativesEP3335699A12016-12-152018-06-20H e x a l AktiengesellschaftSelexipag formulation in liquisolid system
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
475086-01-2  |
|
| ChEMBL | ChEMBL238804  |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
//////////ACT-333679, MRE-269, Selexipag, セレキシパグ , UNII-5EXC0E384L, селексипаг , سيليكسيباق , Orphan Drug, fda 2015, NS 304, ACT 293987, Uptravi, EU 2016,
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3