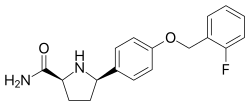
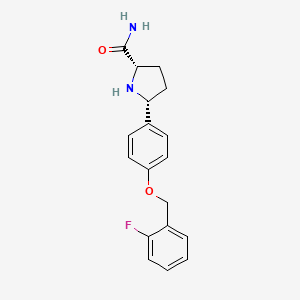
VIXOTRIGINE
- Molecular FormulaC18H19FN2O2
- Average mass314.354 Da
- раксатригин , راكساتريجين , 维索曲静 ,
Vixotrigine (INN, USAN), formerly known as raxatrigine (INN, USAN), is an analgesic which is under development by Convergence Pharmaceuticals for the treatment of lumbosacral radiculopathy (sciatica) and trigeminal neuralgia (TGN).[1][2][3] Vixotrigine was originally claimed to be a selective central Nav1.3 blocker, but was subsequently redefined as a selective peripheral Nav1.7 blocker.[4]Following this, vixotrigine was redefined once again, as a non-selective voltage-gated sodium channel blocker.[4] As of January 2018, it is in phase III clinical trials for trigeminal neuralgia and is in phase II clinical studies for erythromelalgia and neuropathic pain.[5] It was previously under investigation for the treatment of bipolar disorder, but development for this indication was discontinued.[5]
WO2018085521 , claiming novel dosage regimen, assigned to Biogen Inc and Biogen Ma Inc , naming a different team. Biogen, following the acquisition of Convergence Pharmaceuticals , that previously acquired clinical assets from GlaxoSmithKline , is developing vixotrigine ( phase 2 , in November 2018), a voltage-gated sodium channel 1.7 inhibitor, for treating neuropathic pain associated with trigeminal neuralgia, and small fibre neuropathy
PATENT
WO 2011/029762.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011029762
Preparation 1 : Methyl 4-(2-fluorobenzyloxy)benzoate (P1)
Methylparaben (8.85 g, 58.19 mmol) and K2CO3 (16.1 g, 1 16.38 mmol) were stirred in acetonitrile (100 mL) for 5 minutes and then 2-fluorobenzyl bromide (10 g, 52.9 mmol) was added. The suspension was heated to 50-55 °C and held for 2 hours. The mixture was then cooled to 20-25 °C, filtered, and the filtrate solution concentrated to a thick residue. The residue was then dissolved in CH2CI2, washed with a 1 M Na2CO3 solution, dried over Na2SO4, and concentrated to a solid. The solid was then stirred vigorously for 1 hour in just enough hexanes to allow for agitation (~40 mL) and then cooled to 0-5 °C. After 15 minutes, the product was isolated by filtration and washed with -25 mL of hexanes. After drying under vacuum, 1 was isolated as a white solid (13.1 g, 87% yield).
1H NMR (400 MHz, DMSO-d6) δ 7.96-7.90 (2H, m), 7.57 (2H, apparent td, J = 7.7, 1.8 Hz),
7.48-7.39 (1 H, m), 7.30-7.21 (2H, m), 7.17-7.12 (2H, m), 5.22 (2H, s), 3.81 (3H, s).
13C NMR (100 MHz, DMSO-d6) δ 166.2, 162.4, 160.8 (d, J = 247 Hz), 131.6, 131.1 (d, J = 3.8
Hz), 131.0 (d, J = 8.3 Hz), 124.9 (d, J = 3.4 Hz), 123.5 (d, J = 14.1 Hz), 122.6, 1 15.8 (d, J =
21.0 Hz), 115.0, 64.2 (d, J = 3.4 Hz), 52.2.
LRMS (m/e) : 261.3 [MH]+.
Preparation 2: 4-(2-fluorobenzyloxy)benzoic acid (P2).
Methyl 4-(2-fluorobenzyloxy)benzoate (P1 , 10.0 g, 26.9 mmol) was dissolved in methanol (60 mL) and THF (90 mL). A 45 wt% potassium hydroxide solution (20 mL) was then added and
the resulting exotherm was controlled by a water bath. After 1.5 days at 20-25 °C the solution became a thick suspension. Using a water bath to control the exotherm, 20 mL of concentrated HCl was added. The mixture was then concentrated to remove the THF and methanol and 150 mL water was added. The solid was isolated by filtration and washed with 50 mL water. After drying under vacuum, the title compound was isolated as a white crystalline solid (9.4 g, 99% yield).
1H NMR (400 MHz, DMSO-d6) δ 7.95-7.89 (2H, m), 7.58 (2H, apparent td, J = 7.5, 1.7 Hz), 7.48-7.41 (1 H, m), 7.30-7.22 (2H, m), 7.16-7.10 (2H, m), 5.22 (2H, s).
13C NMR (100 MHz, DMSO-d6) δ 167.3, 162.1 , 160.8 (d, J = 246 Hz), 131.7, 131.2 (d, J = 3.8 Hz), 131.0 (d, J = 8.3 Hz), 124.9 (d, J = 3.4 Hz), 123.8, 123.6, 115.8 (d, J = 21.0 Hz), 114.9, 64.2 (d, J = 3.4 Hz).
LRMS (m/e) 247.2 [MH]+.
Preparation 3: 4-(2-fluorobenzyloxy)-N-methyl-N-methoxybenzamide (P3).
4-(2-fluorobenzyloxy)benzoic acid (P2, 5.5 g, 22.3 mmol) was suspended in thionyl chloride (16.5 mL) and heated to 65 °C and held for 3 hours during which time the reactor was kept under a slow sweep of nitrogen. The mixture was then concentrated to a thick oil under hi vac to remove all traces of residual thionyl chloride. The residue was then diluted in CH2CI2 (20 mL) and cooled to 0 °C. In a separate flask, a solution of diaza(1 ,3)bicycle[5.4.0]undecane (DBU, 8.0 mL, 8.15 g, 53.52 mmol) and N-methoxy-N-methyl amine hydrochloride (2.61 g, 26.76 mmol) in CH2CI2 (20 mL) was made and slowly added to the solution at 0 °C. After warming to 20-25 °C, the mixture was washed with 1 M HCl and then with a saturated NaHCO3 solution. After drying over Na2SO4, the solution was concentrated to a thick residue. The mixture was then purified by flash column chromatography eluting with 0→ 100% EtOAc/hexanes (gradient). Concentration of the fractions containing the title compound gave an oil that crystallized upon standing (6.0 g, 93% yield).
1H NMR (400 MHz, DMSO-d6) δ 7.66-7.62 (2H, m), 7.58 (2H, apparent td, J = 7.5, 1.7 Hz), 7.48-7.41 (1 H, m), 7.30-7.23 (2H, m), 7.12-7.07 (2H, m), 5.20 (2H, s), 3.55 (3H, s), 3.25 (3H, s).
13C NMR (100 MHz, DMSO-d6) δ 168.9, 168.0 (d, J = 246 Hz), 163.0, 131.2 (d, J = 3.8 Hz), 130.9 (d, J = 8.2 Hz), 130.4, 126.9, 124.9 (d, J = 3.4 Hz), 123.8 (d, J = 14.8 Hz), 115.8 (d, J = 21.0 Hz), 114.4, 64.0 (d, J = 3.8 Hz), 60.9, 33.8.
LRMS (m/e) 290.3 [MH]+.
Preparation 4: 1-(4-[2-fluorobenzyloxy]phenyl)-2-propen-1-one (P4).
4-(2-fluorobenzyloxy)-N-methyl-N-methoxybenzamide (P3, 6.0 g, 20.7 mmol) was dissolved in THF (100 mL) and cooled to -78 °C. A 1.0 M solution of vinyl magnesium bromide in THF (31 mL, 31 mmol) was added and the cold bath was removed. Upon warming to 20-25 °C, the mixture was poured into a vigorously stirred solution of 1 M HCl. The resulting mixture was extracted twice with CH2CI2. The combined organic layers were then washed with 1 M HCl, then with a saturated NaHCO3 solution, dried over Na2SO4, and concentrated to a thick residue. The product was purified by flash column chromatography eluting with 0→ 40% acetone hexanes (gradient). Concentration of the fractions containing 4 gave an oil that crystallized upon standing (4.83 g, 91% yield).
1H NMR (400 MHz, DMSO-d6) δ 8.06-8.01 (2H, m), 7.59 (1 H, apparent td, J = 7.5, 1.7 Hz), 7.48-7.38 (2H, m), 7.30-7.22 (2H, m), 7.21-7.16 (2H, m), 6.32 (1 H, dd, J = 17.0, 2.0 Hz), 5.92 (1 H, dd, J = 10.5, 2.0 Hz), 5.26 (2H, s).
13C NMR (100 MHz, DMSO-d6) δ 188.3, 162.6, 160.8 (d, J = 246 Hz), 132.5, 131.3, 131.2 (d, J = 3.8 Hz), 131.0 (d, J = 8.2 Hz), 130.3, 129.7, 124.9 (d, J = 3.1 Hz), 123.6 (d, J = 14.4 Hz), 115.8 (d, J = 21.0 Hz), 115.2, 64.3 (d, J = 3.4 Hz).
LRMS (m/e) 257.3 [MH]+.
Preparation 6: Ethyl-5-(4-[2-fluorobenzyloxy]phenyl)-3,4-dihydro-2H-pyrrole-2- carboxylate (P5)
(S)-4-lsopropyl-2-[(S)-2-(diphenylphosphino) ferrocen-1-yl]oxazoline (18.8 mg, 0.039 mmol) and Cu(MeCN)4PF6 (14.5 mg, 0.039 mmol) were added to a dried, nitrogen swept reaction vessel. Anhydrous, degassed, BHT inhibited THF (5.0 mL) was then added and the mixture was stirred for 30 minutes at 20-25 °C. The resulting solution was then cooled to -78 °C and a solution of 1-(4-[2-fluorobenzyloxy]phenyl)-2-propen-1-one (P4, 2.0 g, 7.80 mmol) and ethyl N-(diphenylmethylidene)glycinate (2.29 g, 8.58 mmol) in THF (15 mL total volume) was added over 1-2 minutes. After 3-5 minutes, a solution of DBU (5.9 mg, 0.039 mmol) in THF (0.5 mL total volume) was added. The solution was then stirred for 8-12 hours at -78 °C. The reaction mixture was then warmed to 0-5 °C and 1 M H2SO4 (aq., 25 mL) was then added. The reaction mixture was then warmed to 20-25 °C and mixed vigorously for 2 hours. The mixture was then poured into a rapidly stirring solution of NaHCO3 (saturated, enough to bring the pH to≥ 7.0). After 5minut.es of stirring, the mixture was extracted twice with TBME and the organic extracts were pooled, dried over Na2SO4, and concentrated to near dryness. The resulting residue was purified by flash column chromatography eluting with 0→ 40% acetone/hexanes (gradient). Concentration of the fractions containing the title compound gave a crystalline solid (2.23 g, 84% yield).
1H NMR (400 MHz, DMSO-d6) δ 7.85-7.80 (2H, m), 7.58 (1H, apparent td, J = 7.5, 1.7 Hz), 7.47-7.41 (1 H, m), 7.30-7.22 (2H, m), 7.13-7.09 (2H, m), 5.21 (2H, s), 4.82-4.76 (1 H, m), 4.14 (2H, q, J = 7.1 Hz), 3.13-3.02 (1 H, m), 2.98-2.87 (1 H, m), 2.32-2.21 (1 H, m), 2.09-1.98 (1 H, m), 1.22 (3H, t, J = 7.02 Hz).
13C NMR (100 MHz, DMSO-d6) δ 174.8, 173.1 , 160.8 (d, J = 246 Hz), 160.6, 131.1 (d, J = 3.8 Hz), 130.9 (d, J = 8.3 Hz), 130.0, 127.1 , 124.9 (d, J = 3.1 Hz), 123.9 (d, J = 14.4 Hz), 1 15.8 (d, J = 21.0 Hz), 115.0, 74.2, 64.0 (d, J = 3.8 Hz), 60.7, 35.3, 26.6, 14.4.
LRMS (m/e) 342.4 [MH]+.
Preparation 6: 1-{4-[(phenylmethyl)oxy]phenyl}-2-propen-1-one (P6).
1-{4-[(phenylmethyl)oxy]phenyl}-2-propen-1-one may be prepared from N-methyl-N-(methyloxy)-4-[(phenylmethyl)oxy]benzamide using analogous procedures as those described above for the preparation of P4. N-methyl-N-(methyloxy)-4-[(phenylmethyl)oxy]benzamide may be prepared according to procedures known from the literature (Cowart, M. et. al. J. Med. Chem. 2005, 48, 38).
1H NMR (400 MHz, DMSO-d6) δ 8.05-8.00 (2H, m), 7.50-7.32 (6H, m), 7.18-7.14 (2H, m),
6.32 (1 H, dd, J = 16.9, 2.1 Hz), 5.92 (1 H, dd, J = 10.5, 2.1 Hz), 5.23 (2H, s).
13C NMR (100 MHz, DMSO-d6) d 188.3, 162.8, 136.8, 132.5, 131.3, 130.1 , 129.6, 128.9,
128.4, 128.2, 115.3, 69.9.
LRMS (m/e) 239.3 [MH]+.
Praparation 7a and 7b Ethyl (2R)-2-[(diphenylmethylidene)amino]-5-(4-[2-fluorobenzyloxy]phenyl)-5-oxopentanoate (P7a) and Ethyl (2S)-2-[(diphenylmethylidene)amino]-5-(4-[2-fluorobenzyloxy]phenyl)-5-oxopentanoate (P7b).
The Ligand (according to Table 1 below reported, 0.0084 mmol) and Cu(MeCN)4PF6 (3.13 mg, 0.0084 mmol) were added to a dried, nitrogen swept reaction vessel. Anhydrous, degassed, BHT inhibited THF (0.4 mL) was then added and the mixture was stirred for 30 minutes at 20-25 °C. The resulting solution was then cooled to -20 to -21 °C and a solution of 1-{4-[(phenylmethyl)oxy]phenyl}-2-propen-1-one (P6, 100mg, 0.42 mmol) and ethyl N-(diphenylmethylidene)glycinate (123.5 mg, 0.462 mmol) in THF (0.5 mL total volume) was added over 1-2 minutes. After 1-5 minutes, a solution of DBU (1.27 mg, 0.0084 mmol) in THF (0.1 mL total volume) was added. The solution was then stirred for 8-12 hours at -20 to -25 °C. After this time the reactions were complete and an aliquot of each reaction mixture was diluted in 10% iPrOH / hexanes and analyzed by chiral HPLC. An analytically pure sample was obtained by subjecting the concentrated reaction mixture to flash column chromatography eluting with 0→ 40% acetone hexanes (gradient). Concentration of the fractions containing 7a and 7b (94:6) gave a thick syrup (187 mg, 88% yield).
1H NMR (400 MHz, DMSO-d6) δ 7.91-7.86 (2H, m), 7.54-7.32 (13H, m), 7.13-7.07 (4H, m), 5.20 (2H, s), 4.11-4.05 (2H, m), 4.02 (1 H, dd, J = 8.0, 4.8 Hz), 3.01-2.91 (2H, m), 2.27-2.21 (1 H, m), 2.14-2.08 (1 H, m), 1.16 (3H, t, J = 7.2 Hz).
13C NMR (100 MHz, DMSO-d6) δ 197.3, 171.2, 170.0, 162.1 , 138.8, 136.5, 135.6, 130.5, 130.1 , 129.6, 128.7, 128.6, 128.5, 128.2, 128.1 , 128.0, 127.7, 127.3, 114.6, 69.4, 63.8, 60.5, 33.6, 27.7, 14.0.
Example 1: (5R)-5-(4-[2-fluorobenzyioxy]phenyl)-L-prolinamide (E1)
A mixture of 5% Pt/C (Johnson Mathey B102022-5, 100 mg) was added to a solution of Ethyl -5-(4-[2-fluorobenzyloxy]phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate (P5, obtained as above reported, 1.0 g, 2.93 mmol) in ethanol (12 mL). Acetic acid (1.2 ml.) was then added and the reaction vessel was purged with N2 and then H2. The mixture was hydrogenated at 50 psi of H2 at 15-20 °C for at least 2h. Upon completion of the reaction (monitored by H2 uptake), the mixture was filtered through celite, then through a 0.2 μm PTFE filter and concentrated to approximately 1.5 mL. The mixture was diluted with 1 :1 iPrOAc/TBME and washed with a saturated solution of NaHCO3. After concentrating the organics to a thick residual oil (986mg, 98% crude yield; LCMS retention time 2.04 minutes, calculated 344.4 [MH]+, found 344.3 [MH]+), a solution of ammonia in methanol (ca 7 M) was added in two portions (4 mL initially and then 1 mL after ~10 hrs). After the additions were complete, the reaction stirred for at least 24 hrs at 15-20 °C. Upon completion of the reaction, the mixture was concentrated to dryness. The solid was suspended in a mixture of toluene/TBME 1 :1 (~4 mL) at 18-23 °C with vigorous mixing. After 2hrs at 18-23 °C, the mixture was cooled to 0-5 °C and held for 1 hr. The solid was isolated by filtration and washed with TBME (~4 mL). Drying the solid in a vacuum oven at approximately 40 °C gave the title compound as an off white-solid (720 mg, 78% yield from P5).
Analysis of the sample obtained, performed on CHIRALCEL OJ analytical HPLC column (10% iPrOH/hexanes, 1 mL/min, rt), revealed the presence in minor amounts of (5S)-5-(4-{[(2-fluorophenyl)methyl]oxy}phenyl)-D-prolinamide (enantiomer of the title compound); retention times: (5S)-5-(4-{[(2-fluorophenyl)methyl]oxy}phenyl)-D-prolinamide 36.3 min (1.2%), E1 41.8 min (98.8%).
1H NMR (400 MHz, DMSO-d6) δ 7.55 (1 H, apparent td, J = 7.6, 1.6 Hz), 7.45-7.32 (4H, m), 7.29-7.21 (2H, m), 7.14 (1 H, br. s), 7.00-6.95 (2H, m), 5.12 (2H, s), 4.10 (1 H, dd, J = 9.4, 5.8 Hz), 3.56 (1 H, dd, J = 9.4, 4.4 Hz), 2.14-1.96 (2H, m), 1.92-1.82 (1 H, m), 1.47-1.36 (1 H, m). 13C NMR (100 MHz, DMSO-d6) δ 177.1 , 160.3 (d, J = 246 Hz), 157.0, 137.1 , 130.6 (d, J = 3.8 Hz), 130.3 (d, J = 8.3 Hz), 127.6, 124.5 (d, J = 3.4 Hz), 124.0 (d, J = 14.4 Hz), 115.3 (d, J = 21.0 Hz), 114.4, 63.5 (d, J = 3.8 Hz), 61.7, 59.9, 34.1 , 30.4.
Example 2: (5R)-5-(4-[2-fluorobenzyloxy]phenyl)-L-prolinamide hydrochloride (E2)
To a solution of E1 ( 72 mg, 0.23 mmol) in a mixture of ethyl acetate (1.0 ml) and methanol (1.0 ml) was added 4M HCl in 1 ,4-dioxane (57.5 uL, 0.23 mmol) at 0°C. The mixture was stirred for 1.5h and slowly allowed to warm to room temperature. After evaporating the solvent, the residue was triturated with diethyl ether to afford the title compound as a white solid (75 mg, 93% yield).
1H NMR (300 MHz, DMSO-d6) δ 10.89 (1 H, br. s), 8.12 (1 H, s), 8.1 1 (1 H, br. s), 7.73 (1 H, s), 7.60-7.39 (4H, m), 7.30-7.21 (2H, m), 7.13-7.06 (2H, m), 5.18 (2H, s), 4.66-4.56 (1 H, m), 4.36-4.28 (1 H, m), 2.42-1.94 (4H, m).
PATENT
WO-2018213686
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018213686&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of vixotrigine and their anhydrous form or solvates (designated as Forms A-C), processes for their preparation and composition comprising them are claimed.
The hydrochloride salt of (2S, 5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide, herein referred to as the compound of formula (I):
(I)
is described in WO 2007/042239 as having utility in the treatment of diseases and conditions mediated by modulation of use-dependent voltage-gated sodium channels. The synthetic preparation of (2S, 5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide hydrochloride is described in both WO 2007/042239 and WO 2011/029762.
However, there is a need for the development of crystalline forms of such a-carboxamide pyrrolidine derivatives, which have desirable pharmaceutical properties
Example 1 : (5/?)-5-(4-{[(2-Fluorophenyl)methyl]oxy}phenyl)-L-prolinamide hydrochloride (E1 )
. HCI
The compound of Example 1 may be prepared as described in Example 2,
Procedures 1 to 5 of WO 2007/042239.
Example 2: (5 ?)-5-(4-{[(2-Fluorophenyl)methyl]oxy}phenyl)-L-prolinamide hydrochloride Form 1 (Anhydrous A) (E2)
25.0 mg of Example 1 was added to a 3 mL scintillation vial. THF (2.00 mL) was added and the resulting suspension stirred for 10 minutes. The suspension was filtered through a 0.45 μηι PTFE filter and the filtrate vial placed inside a 20 mL scintillation vial. Hexanes (2 mL) were placed in the outer vial, the entire system sealed and stored at room temperature for 3 days, after which time a crop of colorless crystals was evident in the 3 mL vial. One of these crystals was selected for a single crystal X-ray diffraction experiment. Full characterisation is shown in Figures 1 and 2 and Tables 1 and 2 below
References
- Jump up^ Convergence Pharmaceuticals. “CNV1014802 – Convergence Pharmaceuticals”.
- Jump up^ Stephen McMahon; Martin Koltzenburg; Irene Tracey; Dennis C. Turk (1 March 2013). Wall & Melzack’s Textbook of Pain: Expert Consult – Online. Elsevier Health Sciences. p. 508. ISBN 0-7020-5374-0.
- Jump up^ Bagal, Sharan K.; Chapman, Mark L.; Marron, Brian E.; Prime, Rebecca; Ian Storer, R.; Swain, Nigel A. (2014). “Recent progress in sodium channel modulators for pain”. Bioorganic & Medicinal Chemistry Letters. 24 (16): 3690–9. doi:10.1016/j.bmcl.2014.06.038. ISSN 0960-894X. PMID 25060923.
- ^ Jump up to:a b Keppel Hesselink, Jan M. (2017). “Moving targets in sodium channel blocker development: the case of raxatrigine: from a central NaV1.3 blocker via a peripheral NaV1.7 blocker to a less selective sodium channel blocker”. Journal of Medicine and Therapeutics. 1 (1). doi:10.15761/JMT.1000104. ISSN 2399-9799.
- ^ Jump up to:a b https://adisinsight.springer.com/drugs/800027679
External links
Vixotrigine – AdisInsight
|
|
| Clinical data | |
|---|---|
| Synonyms | Raxatrigine; CNV1014802; GSK-1014802; BIIB 074 |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| Chemical and physical data | |
| Formula | C18H19FN2O2 |
| Molar mass | 314.354 g/mol |
| 3D model (JSmol) | |
////////////VIXOTRIGINE, раксатригин , راكساتريجين , 维索曲静 , QQS4J85K6Y, Raxatrigine, UNII:QQS4J85K6Y


















