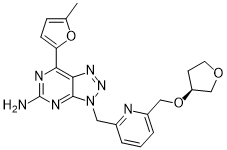

CIFORADENANT
1202402-40-1
Chemical Formula: C20H21N7O3
Molecular Weight: 407.434
CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444,
UNII 8KFO2187CP
Corvus Pharmaceuticals, Inc. PHASE 1
(S)-7-(5-methylfuran-2-yl)-3-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine
| 3H-1,2,3-TRIAZOLO(4,5-D)PYRIMIDIN-5-AMINE, 7-(5-METHYL-2-FURANYL)-3-((6-((((3S)-TETRAHYDRO-3-FURANYL)OXY)METHYL)-2-PYRIDINYL)METHYL)- |
(73 S)-15 -methyl-6-oxa-2(7,3)-[1,2,3]triazolo[4,5- d]pyrimidina-4(2,6)-pyridina-1(2)-furana-7(3)- oxolanaheptaphan-25 -amine adenosine receptor antagonist
Ciforadenant, also known as CPI-444 and V81444, is an orally administered antagonist of the adenosine A2A receptor. Upon oral administration, CPI-444 binds to adenosine A2A receptors expressed on the surface of immune cells, including T-lymphocytes, natural killer (NK) cells, macrophages and dendritic cells (DCs). This prevents tumor-released adenosine from interacting with the A2A receptors on these key immune surveillance cells, thereby abrogating adenosine-induced immunosuppression in the tumor microenvironment.
Ciforadenant is an antagonist of adenosine A2A being developed by Corvus , under license from Vernalis , for the oral treatment of advanced solid tumor; the company is also developing the drug in combination with atezolizumab , for non-small-cell lung cancer.
In 2015, Vernalis licensed the exclusive rights of the product for use of all therapeutic application to Corvus.
Synthesis
WO 2009156737

PATENT
WO 2009156737
US 8450328
WO2017112917
WO 2018175473
WO 2018009972
WO 2018049271
WO 2018022992
PATENT
PATENT
WO-2018183965
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018183965&redirectedID=true
EXAMPLES
Reaction Scheme 1
[0314] Referring to Reaction Scheme 1 , the process to manufacture triazolo[4,5]pyramidine derivatives and intermediates thereof in accordance with the present disclosure, such as the compound known as CPI-444, consists of three chemical steps and uses starting materials known as CP-55, CP-56 and CP-60. The intermediate known as CP-57 is formed at step la without isolation (telescoped) and taken to the next step to form the compound known as CP-58 at step lb. Suzuki coupling using CP-60 during step 2 generates crude CPI-444 which undergoes crystallization during step 3 to form CPI-444.
[0315] Previously described processes for making triazolo[4,5]pyramidine derivatives and intermediates thereof utilized a compound known as CP-59:
[0316] Moreover, such previously described process utilize triethylamine which takes a longer time for the layers to separate where excessive rag layer is observed during phase separation. [0317] The present inventors unexpectedly and surpisingly found that the replacement of CP-59 with CP-60 improved ease of handling and improved process efficiency. In addition, the present inventors unexpectedly and surpisingly found that the use of potassium carbonate (K2CO3) during step 2 improves the phase separation and minimizes rag layer formation upon reaction completion. Finally, Step 3 employs the use of thermocycler in order to facilitate the removal of residual solvents such as isopropyl alcohol.
[0318] Accordingly, the processes in accordance with the teachings of the present disclosure are an improvement over, and are more suitable for commercial scale-up, than processes previously described.
[0319] Starting material (C-55) is commercially available through Astatech, Inc., Keystone Business Park, 2525 Pearl Buck Road, Bristol, PA, 19007, USA; or Suven, SDE Serene Chambers, Road No.5, Avenue 7 Banjara Hills, Hyderabad, 500034, India.
[0320] CP-60 is commercially available through ARK Pharma, Inc., 3860 North Ventura Drive, Arlington Heights, IL, 60004, USA; or Boron Technology Institute, Road No. 2, Building No. 10, room No. 259, Haidian District, Beijing, China.
EXAMPLE 1. Preparation of CP-56
Reaction Scheme 1
Boc20, CbzCI
[0321] Preparation of Dimethyl pyridine-2,6-dicarboxylate:
Pyridine-2,6-dicarboxylic acid (900g, leq) is suspended in methanol(5 volume) and added H2SO4. (19g). The mixture is heated to reflux for approximately 4hr. After reaction completion, the mixture is cooled to 5- 10°C to allow the solids to precipitate. The solids are stirred for an additional hour. The solids are collected by filtration. The wet-cake is re-dissolved in DCM (3 volume) and extract in sequence with an aqueous saturated solution of NaHC03 (2 Volume) followed by with a 5% brine solution (2 Volume). The organic layer is concentrated to dryness to obtain dimethyl pyridine-2,6-dicarboxylate; 914.85g, purity 100%, yield 87.%.
[0322] Preparation of pyridine-2,6-diyldimethanol:
Dimethyl pyridine-2,6-dicarboxylate (885g, leq) is dissolved in EtOH (4425g, 5 Volume) at room temperature. The NaBH4 (341 g, 2eq) is added slowly to the reaction while keeping the internal temperature below 30°C using an ice bath. The reaction is heated to 35°C for approximately 2hrs. After reaction completion, the mixture is cooled to room temperature and adjusted with 32% HCl solution to pH value of approximately 2.5. The mixture is stirred for
2hrs to allow the solids to precipitate. The mixture is then adjusted pH value of approximately 9 using 30% NaOH solution while maintaining an internal temperature below 30°C and stirred at room temperature for about 30 min. The solids are removed by filtration. The filtrate is concentrated at 50°C. The concentrated residual is suspended with isopropanol (4160g, 8 vol)
/water (416g, 0.8 vol) and heated to 70°C for about lhr. The solution is then cooled to room
temperature and stirred for 2hr before cooling to 5-10°C for 30min. The un-dissolved solids are
removed by filtration. The filtrate is concentrated at 50°C. The concentrated residue is charged
with dichloromefhane (2700g, 5vol) and heated to 40 °C for 30min. The suspension is cooled to 5-
10°C and stirred for 30mins. The solid is collected by filtration and dried under vacuum at 40°C to obtain pyridine-2,6-diyldimethanol; 540.77g, purity 100%, yield 85.86%.
[0323] Preparation of 2,6-6 s(chloromethyl)pyridine:
2,6-bis(chloromethyl)pyridine (400g, leq) is suspended in DCM (2000g) and then cooled to 10- 15°C. Thionyl chloride (SOCb; 775g, 3eq) is charged with CH2CI2 (775g) and then added drop- wised into the reaction vessel while maintaining the internal temperature below 20 °C. The reaction is then warmed to room temperature and held for approximately 2hrs. After reaction completion, the 15% aqueous solution of a2C03 (9038g) is pre-cooled to 10-15°C before charging the reaction mixture into the carbonate solution while maintaining internal temperature below 20 °C. The mixture is stirred until gas-evolution is no longer observed. The organic layer is extracted with water (2 x 3200g) and then concentrated at 50°C to a crude product. The concentrated crude is purified by recrystallization using heptane (946g). The mixture is cooled to 5-10°C for 30min. The solid is collected by filtration and wet-cake is washed with heptane and dried at 40°C under vacuum to obtain 2,6-6zs(chloromethyl)pyridine; 442.6g, purity 100%, yield 87.0%.
[0324] Preparation of (3r,5r,7r)-l-((6-(chloromethyl)pyridin-2-yl)methyl)-l,3,5,7-tetraazaadamantan-l-ium:
2,6-to(chloromethyl)pyridine (420g, leq) is dissolved in CH2CI2 (8400g), HMTA (336g, leq) is added into the reaction vessel. The reaction is heated to approximately 40 °C for about 3hrs. Additional HMTA (168g, 0.5eq) is added into the reaction mixture and stirred overnight at room
temperature. The product is collected by filtration. The wet-cake is washed with CthCkand dried under vacuumat 50°C to obtain (3r,5r,7r)-l -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3, 5,7-tetraazaadamantan- 1 -ium; 730g, purity 97.01%, yield 96.58%.
[0325] Preparation of (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride:
(3r,5r,7r)- 1 -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3 ,5 ,7-tetraazaadamantan- 1 -ium (730g, leq) is suspended in EtOH (4380g) before charging 37% HC1 (159g). The mixture is heated to approximately 60 °C for about lhr. After reaction completion, it is cooled to 25°C. MTBE
(1200g) is charged into the suspension. The suspension is then stirred for about 30 min and cooled to 5-10°C for about lhr. The solids are collected by filtration and washed with MTBE and dried at 50°C under vacuum to obtain (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride; 449.56g (after assay correction), purity 98.15%, yield85.23%.
[0326] Preparation of tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate:
(6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride [422.56g (after assay correction), leq] is dissolved in CH2CI2 (5600g) and pre-cooled to 10-15°C. K2CO3 (1632g) pre-dissolved in water (4000g) is charged into the reaction solution solution. The mixture is stirred for about lOmin and then cooled to 10-15°C. Boc-anhydride (603g) is pre-dissolved in CH2CI2 (1808g) before charging into the reactor. The mixture is warmed to room temperature and held for about an hour. After reaction completion, the organic layer is extracted with water (4000g), The organic layer is concentrated to dryness at 50 °C to obtain tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate; 382.93g [after assay correction); purity 99.01%; yield 81%].
[0327] Preparation of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate:
tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamat [ 382.93g (after assay correction) , leq] is dissolved in THF (1 150) and Nal (720g) is added, the reaction is at room temperature for approximately 4hr. After reaction completion, excess Nal and NaCl are filtered off and the filtrate is concentrated at 40°C. The concentrated residue is re-dissolved in ethyl acetate (2300g) and extracted with water (2900g), the organic layer is washed with 10% aqueous solution of Na2S203 (2600g) followed by 5% brine solution (2900g). The organic layer is concentrated to a residue. The residue is re-dissolved in ethyl acetate (4200g), and then filtered. The filtrate is oncentrated and taken up in ethyl acetate (765g) and stirred at room temperature for about 2hr before slowly adding heptane (380g). The solids are filtered and dried at 50°C under vacuum to
obtain tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate; 440g; purity 100%, Yield 85%.
[0328] Preparation of tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate:
A solution of t-BuOK (113g in THF (1.1 kg) is pre-cooled to 5- 10°C, before charging asolutionof (S)-tetrahydrofuran-3-ol (166g) in THF (220g). The mixture is stirred at room temperature for about lhr. A solution of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate (440g, leq) in THF (880g) is pre-cooled to 10-15°C before. The tetrahydrofuranyl solution is slowly charged into reaction solution while maintaining an internal temperature below 1 °C. After about 1 hour another solution of pre-cooled solution of t-BuOK (50g) and (S)-tetrahydrofuran-3-ol (66g) in THF (405g) kg) is slowly added into reaction mixture while maintaining internal temperature below 10 °C. The mixture is stirred at about 10 °C for approximately 1 hour. After reaction completion, the mixture is quenched with water (2200g) and extracted with toluene (4400g). The organic layer is washed with 5% brine (2x 2200g). The organic layer is concentrated to dryness at 50°C under vacuum to obtain tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate; 389g, purity 89.63%, yield 105%.
[0329] Preparation of CP-56 free base:
tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate (389g, leq) is dissolved in CH2CI2 (1556g) and pre-cooled to 0-5°C before charging drop-wise methanesulfonic acid ( MSA; 600g) into the reaction solution while maintaining internal temperature below 20°C. The mixture is warmed to room temperature and hold for about lhr. After reaction completion, water (389g) is added and cooled to 5-10°C. 30% NaOH is charged to adjust the reactor pH to approximately 12.5. The mixture is stirred for about 30 min before extracting with CH2CI2 (1556g). The organic layer is collected and extracted with an aqueous saturated solution of brine (584g). The organic layer is concentrated under vacuum. The residue is re-dissolved in toluene (1560g andthenconcentrated. The concentrated residue is re-dissolved in toluene (1560g) and then filtered. The filtrate is concentrated to dryness at 50°C under vacuum to obtain CP-56 free base; 221g (after assay correction), purity 91%, yield 84.23%.
[0330] Preparation of CP-56:
CP-56 free base (22 lg (after assay correction), leq) is dissolved in MeOH (260g) and EtOH (1300g) and then cooled about 15°C. Oxalic acid (47), pre-dissolved in MeOH (1 lOg is charged into reaction mixture. The reaction is at 15-20°C for 3hr. The mixture is cooled to 0-5°C and
stirred for about an Ihr. The solid is collected by filtration and the wet-cake is washed with EtOH (390g). The solid is dried under vacuum at 50°C to obtain CP-56 crude. Crude CP-56 is re-crystallized from isopropanol (865g) and H20 (lOOg). The mixture is heated to about 70°C to obtain a solution. The solution is slowly cooled to 50°C for Ihr. The mixture is cooled to 0-5°C for about another Ihr. The solid is filtered and washed with isopropanol. The wet-cake is dried at 50°C under vacuum to obtain CP-56; 164g, purity 99%, yield 95%.
[0331] Alternatively, CP-56 can be formed using the following process:
Reaction Scheme 2
7 8 9
[0332] Preparation of Dimethyl pyridine-2,6-dicarboxylate (compound 2):
Charge diacid (1; 628g) into reactor containing methanol (2Kg) and heat to reflux. After reaction completion the reaction is cooled to 30 C and stirred. The wet-cake is filtered and washed with methanol (500g). The wet-cake is dried under vacuum at about 55 °C to obtain diester (680 g, purity >99%; yield 85%).
[0333] Preparation of 6-(hydroxymethyl)picolinamide (compound 4):
Charge diester (2; 600 g) into reactor containing methanol (1.8 kg) and tetrahydrofuran (1.2 kg). Charge slowly sodium borohydride ( aBH4; about 130 g) into the reaction solution while maintaining an internal temperature below 30 °C. After reaction completion aqueous hydrochloric acid (about 350 g of 32% HC1) is charged into the reaction solution. The mixture is concentrated and then charged with dichloromethane (1.8 kg). The organic solution is extracted with water (600 g) and then concentrated to obtain the crude product (3). Crude 3 was dissolved in methanol (1.3 kg) and then charge ammonium hydroxide (20%; 1.3 kg). The solution was stirred until reaction completion before concentrating solution. The residue was taken up in water (600g) and heated to about 60 °C before cooling to 0 °C. The wet-cake was filtered, washed with water and dried in vacuum oven to obtain 6-(hydroxymethyl)picolinamide (about 220 g, >99% purity).
[0334] Preparation of 6-(chloromethyl)picolinonitrile (compound 5):
Charge 6-(hydroxymethyl)picolinamide (about 220 g) into a rector containing acetonitrile (450 g). Charge POCb (519 g and agitate at about 70 °C. After reaction completion the solution is
cooled to about 30 °C before slowly charging into a pre-cool (about 10 °C) reactor with water
(305 g). Charge toluene (1.4 kg) to extract the solution mixture. The toluene phase is washed in sequence with 20 % NaOH (600 g), saturated NaHC03 (300 g) and water (300 g). Toluene is concentrated to obtain crude Cl-nitrile, 5. Isopropyl alcohol (400 g) is charged to dissolve the wet-cake at about 45 °C before cooling to about 0 °C. The wet-cake was filtrated and washed with heptane (150 g) and dried in vacuum oven to obtain 6-(chloromethyl)picolinonitrile (180 g; > 99%.
[0335] Preparation of (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7):
Charge Cl-nitrile (180 g) into a rector containing THF (540 g). Charge Nal (185.7 g) to the reactor and stirred at 50 °C. After reaction completion, the reactor is cooled to 0 °C. In another
reactor, charge t-BuOK (145.6 g) and THF (320 g). Add (S)-tetrahydrofuran-3-ol (31 1.9 g) into the reactor while maintaining internal temperature below 50 °Cto deprotonate the alcohol. Stir
until t-BuOK dissolves. Add THF-OK / THF solution into 6-(iodomethyl)picolinonitrile solution (compound 6) while maintaining internal temperature below 10 °C. Stir at room
temperature until reaction completion. Concentrate the solution to remove THF solvent. Add
ethyl acetate (630 g) and wash by water (420 g). Extract water phase by ethyl acetate (630 g). Combine organic layer and concentrate to obtain oil crude 374 g. The residue was distilled under vaccum (P=3~4 torr, internal temperature 174 °C to 188 °C) to obtain (S)-6-
(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7) as an oily product (204g, >96% purity; 74% yield).
[0336] Preparation of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (compound 9):
Charge (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (180 g) into a rector containing MeOH (1620 g). Charge NaOMe (95.3 g) to the reactor and stirred for 30 min at 30 °C until
reaction completion. The methyl (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinimidate solution (compound 8) was transferred to hydrogenation apparatus containing 50% Ni (60 g). Purge with N2 and then increase the H2 pressure. Under H2 pressure of 5 kg / cm2 and temperature of 30 °C until reaction completion. The reaction is filtered through celite. The filtrate is concentrated. Toluene is charged (1kg) and then concentrated. Then add toluene (1000 g) and filter to remove salt by-products. The filtrate was concentrated to obtain the oil residue of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (136 g; 85% yield, assay 80%, >91% purity).
[0337] Preparation of CP-56:
Charge (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (170 g) into a rector containing isopropyl alcohol (600 g). Set internal temperature of 75 °C. In another reactor,
charge oxalic acid (41.1 g) and water (60 g) and heat solution. Add oxalic acid solution into
CP-56 free-base solution. Cool to 30 °C for about 4 hours and agitate. The wet-cake was filtered
and washed with isopropyl alcohol (175 g) and dried under vacuum drying with heat to obtain crude CP-56 (136.2 g). Charge CP-56 crude (123 g) into a rector containing methanol (1295 g). Stir until CP-56 was dissolved completely. Filter through celite to remove insoluble salt. The filtrate is concentrated. Charge isopropyl alcohol (500 g) and water (50 g) to dissolve CP-56 using heat. Cool to about 30 °C for about 3 hours and stir. The wet-cake was filtrated and
washed by isopropyl alcohol (165 g) and dried under vacuum drying with heat to obtain CP-56 (1 13.4 g. purity = >99 %, > 99% ee).
EXAMPLE 4. Preparation of CPI-444
CP-58 CP-60
C15H16CIN702 CPI-444
1H-17BO3
W: 361 .79 MW: 208.06 C20H21N O3
MW: 407.43
[0349] It is to be noted that other Pd coupling reagents can also be used such as Pd(PPh3)4 or Pd(PPh3)2Cl2.
[0350] A solution of CP-58 (30.0 g, 1 equiv.), CP-60 (approximately 20.8 g, 1.2 equiv.), in THF (approximately 180 mL), K2C03 (approximately 17.5 g), Pd(dtbpf)Cl2(approximately 337 mg), and water (approximately 100 mL) were stirred and heated to about 60 °C until reaction completion. The reaction was cooled to about 50 °C and the layers were allowed to separate. The aqueous layer was removed and back extracted with THF (approximately 30 mL). The THF layers were combined and water (approximately 450 ml) was added to precipitate out crude CPI-444. The slurry was cooled to about 20 °C and stirred for approximately 60 min and the slurry was filtered. The cake was washed in sequence with water (approximately 120 ml) and 2-propanol (approximately 30 ml). The wet-cake was dried in the vacuum oven to provide an off- white solid (29.74 g, 88% yield) with a purity of 98.5 %. Crude CPI-444 conforms to reference.
-444 can be prepared by the following process:
EDA and DAP are used to remove Palladium during CPI-444 formation.
[0352] The solution of CP-58 (10 g), CP-60 (6.9 g) , Pd(dtbpf)C12 (approx. 0.0015 mol eq) and K2C03 (5.8 g) in THF (6V) and H20 (3V) is heated to approximately 60 °C. The reaction is complete after approximately 30 minutes. The solution is cooled to 50 °C and aqueous layer is separated. The aqueous layer is extracted with THF (9 mL); the THF layer is added to organic solution. The organics are cooled to 40 °C, 1 ,3-diaminopropane (DAP; approximately 50 g) or ethylene diamine (EDA; approximately 45 g) is added and the mixture stirred for 1 hour. H20 (15V) is added to the organic layer over 10 min. The slurry is cooled to 20 °C for 2 hours, and stirred for an additional 1 hour. The slurry is filtered and washed with H20 (2V x 2) and z‘PrOH (IV). CPI-444 wet-cake is dried at 50 °C under full vacuum. (Yield = 90 %; purity > 99.0%).
[0353] Alternatively, CPI-444 can be prepared by the following process:
using cysteine in TNF to remove Palladium during CPI-444 formation
[0354] CP-58 (1 kg), K2C03 (0.58 kg), water (3 kg), CP-60 (0.69 kg), and THF (5.3 kg),
Pd(dtbpf)Cb (3 g). The solution is heated to 60 °C. The reaction is complete after approximately 30 minutes. Charge THF (4.5 kg) and cool to 50 °C. The aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. THF is distilled to approximately 7 volumes under atmospheric pressure. The solution is cooled to 50 °C before charging NH4OH (0.5 kg) and agitate for 30 min. Water (14.5 kg) is charged while maintaining the internal temperature >40 °C. The reactor is cooled to 20 °C for 2 hours and hold for an additional 1 hour. CPI-444 is filtered and washed with water followed by isopropanol. CPI-444 wet-cake is dried under vacuum at 50 °C. Purity > 99%, yield 85%.
EXAMPLE 5. Removal of Residual Palladium With Biocap Filter Cartridge
[0355] A mixture of CPI-444 crude (16.00 g), THF (approximately 190 ml), L-cysteine
(approximately 8 g), and H20 (approximately 90 ml) were mixed and heated to a solution at about 60 °C for 1 hour. A solution of 28% NH OH (approximately 20 ml) was added and heated for an additional 15 minutes. The agitation was turned off to allow the layers allowed to settle. The aqueous layer was removed; the THF layer was washed with brine solution (approximately 15 ml). The combined aqueous solutions were back extracted with THF (approximately 15 ml). A 3M Biocap filter (BC0025LR55SP; available from 3M) was pretreated with THF (approximately 150 ml) at about 50 °C. The combined organic layers were recirculated through the Biocap at about 10 ml/min for approximately 3 hours and then filtered forward. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were concentrated. Water
(approximately 80 ml) was added, and distilled to remove residual THF. 2-Propanol (approximately 1 10 ml) was added to the slurry, and the mixture was heated to a solution. The solution was cooled to 20 °C and water (approximately 240 ml) was added. The slurry was performed in series by heating to about 55 °C and held that that temperature for approximately 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was repeated two more. The slurry was then held at 20 °C for approximately 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (about 23 g) was dried in the vacuum oven to obtain an off white solid (13.6 g; 85% yield;99.9% purity; Pd = 25 ppm).
[0356] Reprocess of step 4. AFC-825-106
[0357] CPI-444 (16.02 g, AFC-825-48) and THF (approximately 280 ml) were charged to a flask and heated to about 50 °C for about 30 minutes to obtain a solution. A 3M Biocap filter
(BC0025LR55SP) was pretreated with THF (approximately 150 ml) at about 50 °C . The CPI-444 solution was passed through the Biocap at aboutl O ml/min. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were transferred to a reactor and concentrated. Water (approximately 80 ml) was added, and distilled to remove residual THF solvent. 2-Propanol (approximately 1 10 ml) was added to the slurry and heated to about 65 °C to obtain a solution. The solution was cooled to about 20 °C before adding water (approximately 240 ml). The slurry was heated to 55 °C over 30 minutes, held at 55 °C for 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was two more times. The slurry was then held at 20 °C for 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (26.6 g) was dried in the vacuum oven overnight to obtain 15 as a white solid (95% yield; 99% purity; Pd = 5 ppm).
EXAMPLE 6. Removal of Residual Palladium With Darco KB-G
Crude CPI-444
CPI-444 Drug Substance
[0358] Crude CPI-444 (475 g, 1.17 mol, 1.00 eq), 2-MeTHF (1 1.9 L, 25.0 vol) and WFI water (2.6 L, 5.5 vol) were charged to a 19 L jacketed reactor. The mixture was mechanically agitated under a nitrogen blanket. Nitrogen was bubbled through the solution for 20 minutes. L-Cysteine (242 g, 1.99 mol, 1.71 eq) was then charged. The solution in the reactor was heated to 55±5 °C. Upon reaching 50 °C, the reaction mixture was stirred for 1 hour. 28-30% NH4OH (594 mL, 1.25 vol) was charged via addition funnel, and then the reaction mixture was stirred for 15 min. Agitation was stopped and the reaction was allowed to separate for 1 hour. The aqueous layer was removed. The organic layer was allowed to cool to ambient. The organic layer was filtered and the frit was washed with 2-MeTHF (618 mL, 1.3 vol). The organics were concentrated off by rotary evaporation. WFI water (2.42 L, 5.1 vol) and IPA (2.38 L, 5.0 vol) were used to charge the concentrated slurry to a clean 19 L jacketed reactor under N2. The mixture was heated to 65±5 °C, and then was stirred for 1 hour to obtain solution. Darco KB-G activated carbon (71.3 g, 15 wt%) was charged. The reactor was heated to 75±5 °C and stirred for 15 hours. A I L pocket filter was prepared with filter cloth and a heating jacket and heated to 70±5 °C. Reactor contents were filtered through the pocket filter using N2 pressure. The pocket filter was rinsed with a mixture of IPA/WFI water (1 : 1, 950 mL, 2 vol) followed by a mixture of IPA/WFI water (1 : 1, 1.90 L, 4 vol) and IPA/WFI water ( 1 : 1 , 1.90 L, 4 vol). Inside a 22 L three neck round bottom flask the filtrates were mechanically agitated under a N2 blanket. WFI water (7.13 L, 15 vol) was slowly added via addition funnel over 1 h at ambient temperature, and aged for 1 h. The slurry was heated to 55±5 °C and maintained the temperature for 30 min. This heating and subsequent cooling were repeated twice more. After reaching ambient
temperature the final time, the mixture was stirred for at least 2 hours. The reaction mixture was filtered and the reactor rinsed with WFI water (2.38 L, 5.0 vol, 3x). The cake was dried under N2 for 30 minutes and then transferred to a glass dish. The material was dried under full vacuum at 55±5 °C. The desired product was obtained 368.1 g (77%) as light yellow solids. This material was 99.6% pure by HPLC and had a Pd content of 3.6 ppm.
EXAMPLE 7. Removal of Residual Palladium With Polymer-Bound Thiol (SiST)
[0359] Crude CPI-444 (24.48 g, pd = 1267 ppm) and THF (244.8 mL, 10 vol) were charged to a 500 mL 4-necked flask fitted with mechanical agitation, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and then slowly cooled to 45 °C. SiST (36.72 g) was added to the solution and the mixture was stirred at 42 °C for 14 h. The mixture was filtered and washed by THF (24 mL, 1 vol, twice; Pd= 13.12 ppm). H20 (120 mL, 5 vol) and IPA (120 mL, 5vol) were charged to the flask. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to room temperature and the slurry was added H20 (360 mL, 15 vol) and heated to 55 °C for 1 h. The slurry was cooled to room temperature and then heated to 55 °C for 1 h. The slurry was cooled to rt. and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (100 mL, 4 vol, three times). The wet cake (28.36 g) was dried by 10 mmHg and 50 °C for overnight (14h) and the weight of CPI-444 was 19.31 g (79% recovery).
EXAMPLE 8. Removal of Residual Palladium By Recrystallization
[0360] CUNO Filter Cartridge 55 S
[0361] CPI-444 (5.0 g, Pd 14.06 ppm) and THF (50 mL, 10 vol) were charged to a 100 mL 3-necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and added CUNO 55S filter (0.75 g, 15w%). The mixture was stirred at 60 °C for 1 h. The mixture was filtered and washed by THF (5 mL, 1 vol, twice). The filtrate was concentrated. The solid, H20 (25 mL, 5 vol) and IPA (25 mL, 5vol) were charged to 250 mL 3 -necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to rt.(40 minutes) The slurry was added H20 (75 mL, 15 vol) and then heated to 55 °C for 1 h. The slurry was cooled to rt. (30 minutes) and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (20 mL, 4 vol, three times). The cake (6.355 g) was dried by 10 mmHg and 50 °C
for overnight (16 h) and the weight of CPI-444 was 4.281 g (85% recovery). Pd content(ppm) = 2.02 ppm.
[0362] Polymer-bound Thiol: SiST
[0363] CPI-444(5 g; Pd 14.06ppm) was dissolved in THF (50 mL) at 60 °C. The solution was cooled to 55 °C and SiST (7.5 g) was added to the solution. The solution was stirred at 50-55 °C for 16 h. The solution was filtered through celite and a 0.2 micron filter. The filtrate was tested for Pd content. Result: 2.43 ppm.
Catalyst
Molecular Weight: 291.6990
Molecular Weight: 337.3430
[0364] 1. A solution of S.M., CP-60, Pd(PPh3)2Cl2 and K2C03 in THF – H20 (7.9 mL, 1 : 1) was put in oil-bath at 70-75 °C.
[0365] 2. After 2 h, 0.047 g CP-60 was added to the reaction at 70-75 °C.
[0366] 3. After 1 hr, the reaction was cooled to rt. and 10 mL H20 was added to the reaction.
[0367] 4. The reaction was filtered to provide wet cake (0.812 g).
[0368] 5. The solid wet cake was dried at 45 °C and 20 mmHg for 2h to provide weight 0.499 g. (86%).
[0369] 6. The solid wet cake was stirred in 2 mL DMF for 30 mins (slurry) and then filtered. The solid was dried by 45 °C and 10 mmHg for 12h to provide weight 0.40 g; 69% yield; 98.1% purity.
//////////CIFORADENANT, CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444, UNII 8KFO2187CP, Corvus Pharmaceuticals, Inc., PHASE 1,
NC1=NC2=C(N=NN2CC3=NC(CO[C@H]4CCOC4)=CC=C3)C(C5=CC=C(O5)C)=N1